Metastasis is the process of dissemination of a tumor, whereby cells from the primary site dislodge and find their way to other tissues where secondary tumors establish. Metastasis is the primary cause of death related to cancer. This process warrants changes in original tumoral cells and their microenvironment to establish a metastatic niche. Traditionally, cancer therapy has focused on metastasis prevention by systematic treatments or direct surgical re-sectioning. However, metastasis can still occur. More recently, new therapies direct their attention to targeting cancer stem cells. As they propose, these cells could be the orchestrators of the metastatic niche.
- Metastasis
1. Introduction
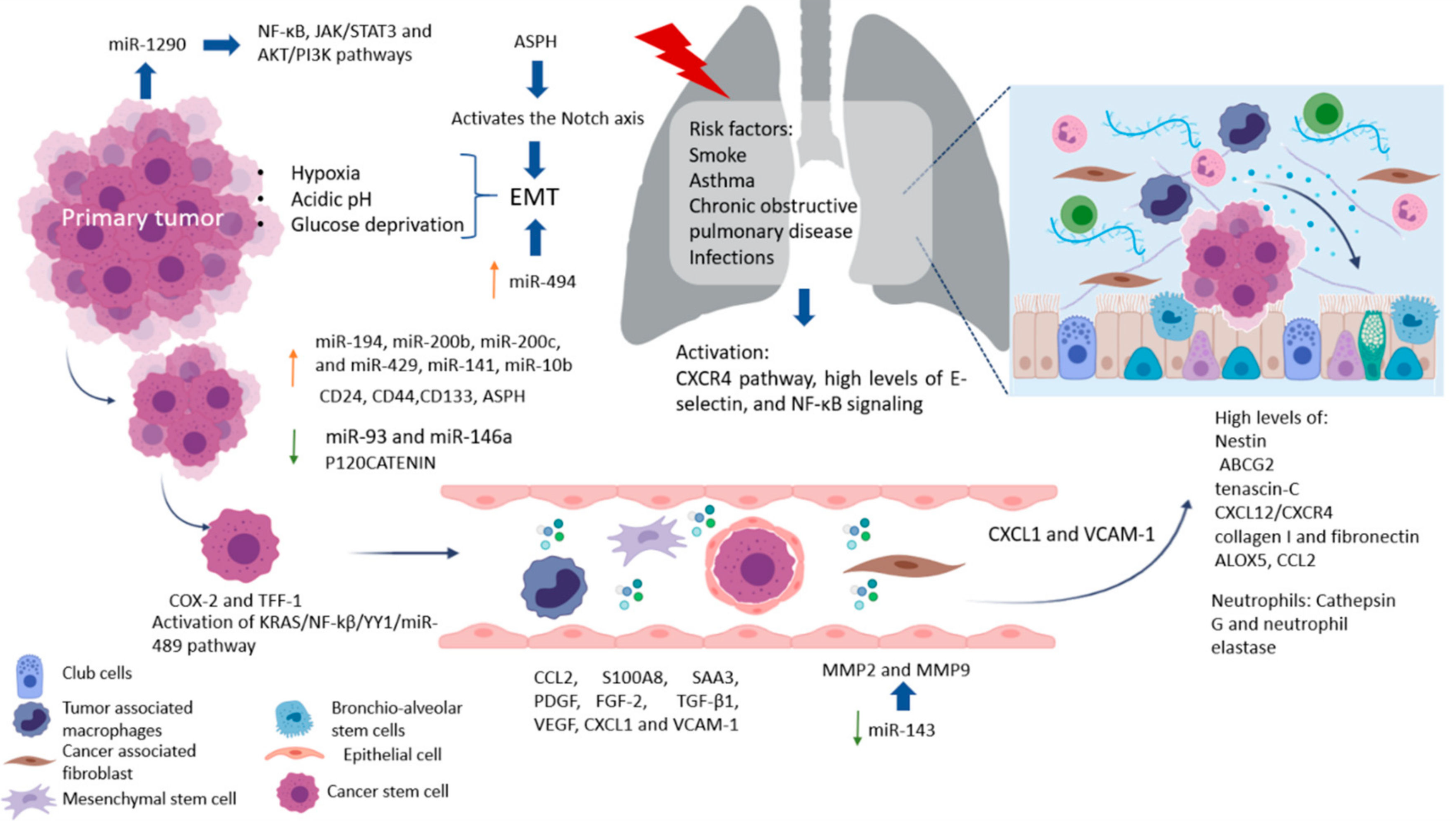
Metastasis is an inefficient process in which cells from the primary tumor spread by releasing circulating tumor cells (CTCs) into vasculature towards a distant organ to colonize it, establishing metastasis. A process where only 0.01% of the cells that enter the circulation can successfully reestablish a new metastasis, and while this seems highly inefficient, it is more often than not a fatal step in cancer progression [1], [2]. Cancer stem cells (CSC) are the subpopulation of cells responsible for promoting angiogenesis, local invasion, distant metastasis, and resistance to apoptosis; moreover, epithelial tumor cells (mature population) gain invasiveness and migratory abilities through the process of epithelial-mesenchymal transition (EMT), which is where a complex network of interconnected factors and pathways meet, such as transforming growth factor-β (TGFβ), epidermal growth factor (EGF), insulin-like growth factor (IGF), WNT, Hedgehog, and Notch pathways, all of which regulate and promote CSC growth [3]–[5].
Metastasis is an inefficient process in which cells from the primary tumor spread by releasing circulating tumor cells (CTCs) into the vasculature towards a distant organ to colonize it, establishing metastasis. A process where only 0.01% of the cells that enter the circulation can successfully reestablish a new metastasis, and while this seems highly inefficient, it is more often than not a fatal step in cancer progression [1][2]. Cancer stem cells (CSC) are the subpopulation of cells responsible for promoting angiogenesis, local invasion, distant metastasis, and resistance to apoptosis. Moreover, epithelial tumor cells (mature population) gain invasiveness and migratory abilities through the process of epithelial–mesenchymal transition (EMT), which is where a complex network of interconnected factors and pathways meet, such as transforming growth factor-β (TGFβ), epidermal growth factor (EGF), insulin-like growth factor (IGF), WNT, Hedgehog, and Notch pathways, all of which regulate and promote CSC growth [3][4][5]. The spread of CSC from the primary tumor to a secondary site is a process highly dependent on signaling cues, such as hypoxia, acidic pH, and/or glucose deprivation [6]. However, only one out of 500 CSC will survive in the circulation, even though mechanisms to protect CSC from elimination by the immune system exist as the secretion of IL-4 and CD200, which represents an important role in immune escape [7]. CSC in the bloodstream or otherwise in the lymphatic system can cluster together with stromal cells (fibroblasts, endothelial, tumor-infiltrated myeloid cells, or pericytes) for improved metastasis potential. Endothelial cells (ECs) in healthy established vessels remain quiescent for years. Under certain conditions, such as hipoxia or inflammation, as occurs in pathologies such as cancer and wounds, they can rapidly switch to an angiogenic state and start to form new blood vessels by interaction with pericytes [8][9][10]. Once an angiogenic switch is turned on, factors (VEGF, PDGF, TNF-α, and IL-8) inside the tumor promote growth and metastasis [11][12]. A majority of ECs in the tumor vasculature are tumor-derived ECs (TECs) retain remain distinct from cancer cells, by are not immortal and their ontological endothelial identity permit participate in making up the lining of neoangiogenic vasculatures in the TME and accelerating tumor progression [13][14]. Circulating tumor endothelial cells (CTECs) with a mature phenotype, derived from vessel wall turnover, play an important role in tumor initiation, progression, metastasis, and neovascularization [15]. Moreover, it has been found clusters of endothelial cells expressing endothelial markers as vimentin and other lineage markers, such as FN1, SERPINE1, and FOXC1, that improve the survival of cancer stem cells and promote their dissemination [15]. Increased CTECs in cancer patients have a worse prognosis, indicated as potential biomarkers of angiogenesis and metastasis, and can express PDL-1 that permit the evaluation of immunotherapy efficacy [16].
The spread of CSC from the primary tumor to a secondary site is a process highly dependent on signaling cues, such as hypoxia, acidic pH, and/or glucose deprivation [6]. CSC in the bloodstream or otherwise in the lymphatic system can cluster together with stromal cells (fibroblasts, endothelial, tumor-infiltrated myeloid cells, or pericytes) for improved metastasis potential. Once CSC attaches and develops with success, a pre-metastatic niche will form. Pre-metastatic niches additionally require exosomes or exosomal-like extracellular vesicles (ECV) from entrained bone marrow-derived cells and macrophages to start changes in the cells. Ideally building an effective microenvironment that serves as a fertile niche for tumor cell growth [8]–[10].
Researchers estimate 20-54% of malignant tumors develop metastasis. Primarily at lymph nodes, with the liver and lungs as the second most common sites of metastasis [11]. Colorectal cancer, the 3rd most frequent neoplasm, can progress to the liver (40-50%) and lung (10-20%) depending on the primary tumor's stage. Meanwhile, pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers with high metastatic potential which caused metastatic disease 19–39% in the lungs and >50% in the liver. Interestingly, several studies have reported isolated lung metastases associate with better survival outcomes than patients with other solitary metastatic organs [14]–[16]. In the following, we will discuss differences between the cascade of events in primary tumors which promote the establishment of pre-niche and niche metastasis.
Why Liver Metastasis?
The physiological functions of the liver are related to the metabolism of nutrients absorbed by the gastrointestinal tract and the detoxification of dangerous molecules present in the circulation. These functions are responsible for a delicate balance between immune tolerance and immune response. Their correct functioning requires normal cytoarchitecture of the hepatic lobules [17].
Due to the total blood volume that circulates through the liver, about 30% of blood volume per minute, it is the preferred organ for metastasis development [18]. The portal vein is a vessel that carries blood from the gastrointestinal tract, gallbladder, pancreas, and spleen to the liver. The right portal vein is the continuation of the main portal vein, while it separates the left portal vein from the main portal vein at an acute angle [19]; which explains why the segment of the right lobe liver is the main target of metastases. Also, the blood circulation in the colon and proximal rectum drain through the hepatic portal system, while the blood of the distal rectum goes to the lung. This vascular organization correlates with the fact that colorectal cancer prefers liver metastasis, with the lung as the second favored metastatic site [20].
However, to allow hepatic metastasis development, the liver must present damage. There are several hypotheses regarding the pro-metastatic state consequent to cirrhosis, steatosis, and nonalcoholic fatty liver disease. Some authors state that altered hepatic cytoarchitecture creates an unfavorable environment [21]. Others report that these alterations slow the passage of neoplastic cells through the microcirculation and enhance cellular-vascular contact and stress micro thrombotic phenomena through the expression of adhesion molecules, facilitating metastasis[22]. Chronic inflammation that results from receiving nutrients, toxins, and microorganisms via the portal vein from the gut can leads to liver injury creating a microenvironment favorable for the increased growth of metastases [21].
Factors crucial for the formation of liver metastasis
Pre-niche is formed by the preconditions in specific organs in terms of nutrients, extracellular matrix, and immune cells necessary to generate a fertile soil before the arrival of CTC, which increases the success of metastasis establishing [24]. After that, primary tumors can induce stromal cell remodeling of the extracellular matrix which takes care of cell organization. To increase the survival of CTC from the hostile microenvironment, they usually form CTC clusters. Interactions between cancer cells and endothelial cells with an endothelial layer, and the underlying basement membrane VCAM, ICAM, or L1 molecules, expressed by the vascular endothelium, interaction with α4β1 integrin and αvβ3 integrin, play a central role in leukocyte recruitment to develop this process [22], [25].
Other tumor-recruited cells such as cancer-associated fibroblasts (CAFs), stromal myofibroblast, endothelial cells, pericytes, diverse immune cells, mesenchymal stem cells (MSCs), and tumor-associated macrophages (TAMs) contribute to tumor growth and serve as a prerequisite for tumor cell invasion and metastasis [8], [24]. For example, during the early stages of tumor development, cytotoxic immune cells such as natural killer (NK) and CD8+ T cells recognize and eliminate the more immunogenic cancer cells. High levels of tumor-infiltrated T cells indicate a good prognosis in many solid tumors, while high levels of macrophage infiltration correlate with a worse prognosis. TAMs promote tumor progression in different ways, such as secreting cytokines like IL-10 and TGF-β, which induce immunosuppression and impair poor response by cytotoxic lymphocytes T and dendritic cell maturation. Also, TAMs release a plethora of extracellular matrix (ECM) remodeling factors (plasminogen activation system, matrix metalloproteinases, and kallikrein-related peptidases), affecting the composition, structure, and elasticity of the ECM and the availability of growth factors, creating conduits for the migration of tumor cells [26].
Metastasis-associated macrophages (MAMs), in murine CRC models, have been shown to increase the extravasation of tumor cells and help in their survival by secreting growth factors and concomitantly inhibiting cytotoxic T cells [27]. Some key mediators in regulating immune responses are myeloid-derived suppressor cells (MDSC) which express CD11b+ and CD33+. CD11b is a subunit of the integrin adhesion molecule, which expresses in bone marrow-derived immune cells. CD11b was reported to promote myeloid cell migration to the tumor microenvironment, which secreted cytokines related to tumor growth and angiogenesis. Researchers relate the expansion of MDSCs to tumorigenesis in CRC, and the density of CD33+ MDSCs in the microenvironment was a negative prognostic factor in CRC patients. Recently reported higher expression (lymphocytes CD3+) in primary tumor than in hepatic metastases, while CD33 had higher expression in hepatic metastases than in primary tumor. In turn, CD33+ showed that more immunosuppression of cells in the liver may contribute to the poor response to immunotherapy [28]–[30]. Although to date, some cell types have been associated with better or worse prognoses of primary tumors, unlike in metastasis where there are many other factors involved, such as the treatment they are receiving; this may change the microenvironment and therefore favor the progression of the disease. In most patients, physicians do not focus the treatment used on destroying the cells responsible for tumor chemoresistance and progression, therefore this approach is being increasingly included in patients with already advanced stages, increasing their survival.
Factors involved in tumor progression and the development of metastasis is hypoxia, which in CRC is very common and becomes more aggressive, invasive, and resistant to chemo‐ and radiotherapy. It associates with the activation of transcription factors involved in the maintenance of EMT/CSC phenotypes and enrichment of CD44high, CD24low or ALDHhigh CSC population [37], [38]. A recent review by Gonzalez-Villareal in colorectal cancer metastasis emphasized EMT regulation, with members of the miR-200 family [24].
In liver metastasis of CRC, researchers observed an enhancement of fructose metabolism via the upregulation of enzyme aldolase B (ALDOB), hence providing extra fuel for metastatic outgrowth [49]. Thus, as the metabolic center of the entire organism, the liver appears to provide a unique milieu enabling or forcing cancer cells to assume specific metabolic activities for colonization. It involves molecular pathways for liver colonization, including nitric oxide and ROS, and the expression of adhesion molecules such as selectins and integrins. Also involved are phagocytosis, cytokines, such as TNFα and TGFβ, interferon gamma (IFNγ), interleukins (IL-1, IL6, IL8, IL-10, IL12, and IL18), growth factor monocyte chemoattract protein-1 (MCP-1), and macrophage inflammatory protein (MIP-1) released by KCs and HSCs [50]. Proinflammatory cytokines such as IL-6, preparing a permissive inflammatory pre-metastatic niche where circulating CRC cells can home and survive, colonize, and eventually form micro and macro metastasis. IL-6 then activates signal transducer and activator of transcription 3 (STAT3) signaling in hepatocytes, which produces serum amyloid A protein (SAA) that orchestrates the formation of a pro-metastatic niche in the liver. It has been reported that S1PR1–STAT3 upregulation in tumor cells induces IL-6, which activates S1PR1–STAT3 in MDSCs in the liver, leading to pre-metastatic niche formation prior to CRC cell arrival [51].
Expression of fibronectin and granulin by macrophages stimulates HSC for differentiation into myofibroblasts, whose release periostin, creates a fibrotic environment in the liver that sustains tumor growth[52]. Similarly, human hepatic sinusoidal endothelial cells in vitro express macrophage migration inhibitory factor (MIF-1), which improves EMT, migration, proliferation, and apoptotic resistance in CRC cells. Finally, angiopoietin-like 6 protein from the liver sinusoidal endothelial cells (LSEC) induces liver colonization of CRC cells and correlates with CRC progression in in vitro models [53].
Recent works reported that type XII collagen was the most significantly upregulated collagen in cancer liver metastasis (CLM) and that there was a difference with the ECM of the CRC. Collagens that are present in metastatic disease were COL10A1, COL12A1, COL 14A1, and COL15A1. Also, collagen type IV has a strong association with liver metastasis in CRC.
Some factors influencing liver metastasis on CRC were found, but we know that tumor cells themselves, stromal cells, and the interaction between CRC cells and their microenvironment, all contribute to hepatic invasion. Until now, we had known about the establishment of the pre-metastatic niche but little about the metastatic niche itself. Therefore, the search for mechanisms and possible therapeutic targets that can reverse or change the prognosis of the disease is currently being continued since most patients at the time of diagnosis already arrive in very advanced stages of the disease, when the approach to treatment is more complex than just treating the primary tumor and preventing metastasis. Next, the management of patients with liver and lung metastases in two neoplasms that are on the rise without hereditary history will be reviewed.
Once CSC attaches and develops with success, a pre-metastatic niche will form. Pre-metastatic niches additionally require exosomes or exosomal-like extracellular vesicles (ECV) from entrained bone marrow-derived cells and macrophages to start changes in the cells. Ideally building an effective microenvironment that serves as a fertile niche for tumor cell growth [8][9][10]. Researchers estimate that between 20% and 54% of malignant tumors develop metastasis. Primarily at lymph nodes, with the liver and lungs as the second most common sites of metastasis [17]. Colorectal cancer, the third most frequent neoplasm, can progress to the liver (40–50%) and lung (10–20%) depending on the primary tumor’s stage. Clinical data show that median survival is just 5–20 months without treatment [18]. Meanwhile, pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers with high metastatic potential. At the time of PDAC diagnosis, approximately 50% of patients present metastatic disease with 19–39% affected in the lungs and >50% in the liver. The median survival time is around 6–11 months in patients with metastatic pancreatic cancer [19]. Interestingly, several studies have reported that isolated lung metastases are associated with better survival outcomes than patients with other solitary metastatic organs [20][21][22].
2. Lung Metastasis
The lungs are one of the most complex organs in the human body. Their function is to exchange oxygen from the external environment with carbon dioxide from the cardiovascular system. Its role in the development of metastasis can result after physical trauma that induces local inflammation (smokers, asthma, obstructive pulmonary disease, or pneumonia) and tissue damage, creating a favorable environment that attracts metastatic cells from distant sites. Also, it has been reported that lung inflammation in smokers or in people with lung diseases such as asthma, chronic obstructive pulmonary disease, and infections, such as pneumonia, are risk factors for lung metastases. Some factors related to the migration of cells and vascular permeability are CCL2, S100A8, and SAA3. Upregulation of fibronectin colocalized with LOX, and higher levels of expression of VCAM-1, a receptor for VLA-4 in patient samples of metastatic tumor nodules in the lungs and bone, have been found [23]. The pro-metastatic effect played by exposure to smoking is related to the activation of the ubiquitin-chemokine receptor type 4 (CXCR4) pathway, high tissue levels of E-selectin, activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), signaling in pneumocytes, increased chemokine ligand 2 (CCL2) expression, and macrophage infiltration in the lung microenvironment. Moreover, lung alveolar cells induce chemokine secretion, which recruits neutrophils. The latter, through the synthesis of arachidonate 5-lipoxygenase (ALOX5)-dependent leukotriene, may promote survival and proliferation of leukotriene B4-expressing metastatic clones [17]. Inflamed lungs recruit neutrophils to release Cathepsin G and neutrophil elastase and destroy the protein Thrombospondin 1 (Tsp-1), which protects lung tissue from metastasis. Another alternative, but not mutually exclusive, is that micro-metastatic foci are already present at the time of physical injury [24]. Therefore, it is believed that lung metastases depend on circulatory system involvement, which flows into the pulmonary arterial system through the subclavian vein via the thoracic duct from the lymph nodes invaded by tumor cells [25]. More recently, DPC4 loss is shown to be associated with EMT, tumor progression, and the presence pulmonary metastases [26].
Factors Crucial for the Formation of Lung Metastasis
Factors produced by cells in pre-niche can support survival and growth of disseminated tumor cells were in Figure 1. The interstitial space is the main site for lung metastasis, which is adjacent to the terminal bronchioles. Bronchioalveolar stem cells (BASC) are situated in the terminal bronchioles and present pro-surfactant apoprotein-C (SP-C), a marker for alveolar type II (AT2) cells, and CC10, a marker for club cells—both are related with the lung pre-metastatic niche. Bone marrow cells, club cells, and alveolar macrophages are also present in pre-metastatic lung [23].
References
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Kuşoğlu, A.; Avcı, Ç.B. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85.
- Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, J.B.; Hermann, P.C. EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136.
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672.
- Mendoza, M.; Khanna, C. Revisiting the seed and soil in cancer metastasis. Int. J. Biochem. Cell Biol. 2009, 41, 1452–1462.
- Codony-Servat, J.; Rosell, R. Cancer stem cells and immunoresistance: Clinical implications and solutions. Transl. Lung Cancer Res. 2015, 4, 689–703.
- Garza Treviño, E.N.; González, P.D.; Valencia Salgado, C.I.; Martinez Garza, A. Effects of pericytes and colon cancer stem cells in the tumor microenvironment. Cancer Cell Int. 2019, 19, 1–12.
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238.
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948.
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272.
- Salazar, N.; Zabel, B.A. Support of Tumor Endothelial Cells by Chemokine Receptors. Front. Immunol. 2019, 10, 147.
- Lin, P.P. Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells 2020, 9, 1539.
- Wei, Y.F.; Yang, Q.; Zhang, Y.; Zhao, T.J.; Liu, X.M.; Zhong, J.; Ma, J.; Chen, Y.X.; Zhao, C.; Li, J.X. Plumbagin restrains hepatocellular carcinoma angiogenesis by suppressing the migration and invasion of tumor-derived vascular endothelial cells. Oncotarget 2017, 8, 15230–15241.
- Cima, I.; Kong, S.L.; Sengupta, D.; Tan, I.B.; Phyo, W.M.; Lee, D.; Hu, M.; Iliescu, C.; Alexander, I.; Goh, W.L.; et al. Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci. Transl. Med. 2016, 8, 345ra89.
- Zhang, L.; Zhang, X.; Liu, Y.; Zhang, T.; Wang, Z.; Gu, M.; Li, Y.; Wang, D.D.; Li, W.; Lin, P.P. PD-L1+ aneuploid circulating tumor endothelial cells (CTECs) exhibit resistance to the checkpoint blockade immunotherapy in advanced NSCLC patients. Cancer Lett. 2020, 469, 355–366.
- Stella, G.M.; Kolling, S.; Benvenuti, S.; Bortolotto, C. Lung-seeking metastases. Cancers 2019, 11, 1010.
- Jahanafrooz, Z.; Hashemzaei, M. Colon cancer therapy by focusing on colon cancer stem cells and their tumor microenvironment. J. Cell. Phys. 2019, 235, 4153–4166.
- Kulke, M.H. Metastatic Pancreatic Cancer. Curr. Treat. Options Oncol. 2002, 3, 449–457.
- Oweira, H.; Petrausch, U.; Helbling, D.; Schmidt, J.; Mannhart, M.; Mehrabi, A.; Schöb, O.; Giryes, A.; Decker, M.; Abdel-Rahman, O. Prognostic value of site-specific metastases in pancreatic adenocarcinoma: A Surveillance Epidemiology and End Results database analysis. World J. Gastroenterol. 2017, 23, 1872–1880.
- Bellon, E.; Gebauer, F.; Tachezy, M.; Izbicki, J.R.; Bockhorn, M. Pancreatic cancer and liver metastases: State of the art. Update Surg. 2016, 68, 247–251.
- Deeb, A.; Haque, S.-U.; Olowokure, O. Pulmonary metastases in pancreatic cancer, is there a survival influence? J. Gastrointest. Oncol. 2015, 6, E48–E51.
- Maru, Y. The lung metastatic niche. J. Mol. Med. 2015, 93, 1185–1192.
- El Rayes, T.; Catena, R.; Lee, S.; Stawowczyk, M.; Joshi, N.; Fischbach, C.; Powell, C.A.; Dannenberg, A.J.; Altorki, N.K.; Gao, D.; et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc. Natl. Acad. Sci. USA 2015, 112, 16000–16005.
- Font-Clos, F.; Zapperi, S.; La Porta, C.A.M. Blood Flow Contributions to Cancer Metastasis. iScience 2020, 23, 101073.
- Shin, S.H.; Kim, H.J.; Hwang, D.W.; Lee, J.H.; Song, K.B.; Jun, E.; Shim, I.K.; Hong, S.-M.; Kim, H.J.; Park, K.M.; et al. The DPC4/SMAD4 genetic status determines recurrence patterns and treatment outcomes in resected pancreatic ductal adenocarcinoma: A prospective cohort study. Oncotarget 2017, 8, 17945–17959.