Paroxysmal nocturnal hemoglobinuria (PNH) is a rare clonal disease that presents an estimated incidence of 1.3 cases per million per year, with a prevalence of 15.9 cases per million. It is characterized by hemolysis, bone marrow dysfunction with peripheral blood cytopenia, hypercoagulability, thrombosis, renal impairment and arterial and pulmonary hypertension. Hemolysis and subsequent hemosiderin accumulation in tubular epithelium cells induce tubular atrophy and interstitial fibrosis.
- Paroxysmal Nocturnal Hemoglobinuria
- Complement Inhibition
- Thrombosis
- Membrane Attack Complex
- Complement Dysregulation
PNH Genetic Mutation
The cause of PNH is a somatic mutation in the X-linked phosphatidylinositol glycan class A (PIG-A) gene on Xp22 [1], coding for one of the several enzymes involved in the generation of glycosyl phosphatidylinositol (GPI) anchors in the endoplasmic reticulum. Deficiency in PIG A leads to a lack of the expression of approximately 150 cell surface proteins [[2]]. The common core structure of GPI is made up of a molecule of phosphatidylinositol and a glycan core consisting of three mannoses, an ethanolamine phosphate and glucosamine [[3]].
At least 10 reactions and more than 20 different genes are implicated in the biosynthesis of GPI anchors [[4]]. In PNH, this mutation results in the production of clonal blood cells with a deficiency in those surface proteins that protect against damage caused by the complement system [[5]]. In this way, such a disorder makes these cells excessively susceptible to the lytic action of activated complement.
PNH features are closely connected with a deficit in or complete absence of CD55 (a decay accelerating factor) and CD59 (a membrane inhibitor of reactive lysis), from the family of GPI-anchored proteins. These proteins modulate solid phase complement activity; CD55 inhibits alternative pathway C3 and C5 convertases, and CD59 prevents the creation of the membrane attack complex (MAC) [[6]] (Figure 1).
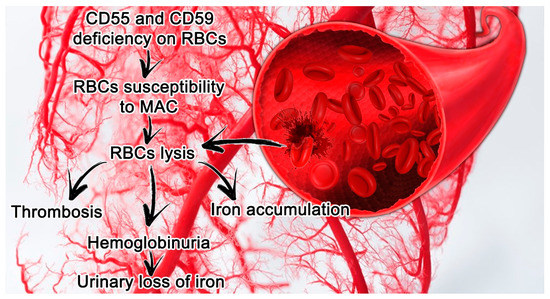
Figure 1.[7] Paroxysmal nocturnal hemoglobinuria (PNH) features in small vessels and its consequences. Erythrocytes lacking CD55 and CD59 are more susceptible to hemolysis mediated by MAC, which leads in turn to thrombosis and the release of hemoglobin and free iron. Abbreviations: CD55, a decay accelerating factor; CD59, a membrane inhibitor of reactive lysis; RBC, red blood cells; MAC, membrane attack complex.
Other proteins have a minor role in PNH pathophysiology, the most common of which are monocyte differentiation antigen (CD14), low-affinity immunoglobulin gamma Fc region receptor III-B (CD16b) and CD48 antigen [[8][9][10]].
PNH Laboratory Findings and Clinical Manifestations
The major clinical manifestations and complications of PNH are associated with hemolytic anemia, thrombosis and bone marrow failure [[11]].
Laboratory results in PNH patients highlight anemia with negative Coombs tests and hemoglobinuria with dipsticks positive for heme but negative sediments for red blood cells, increased reticulocyte counts, elevated levels of lactate dehydrogenase (LDH) and bilirubin, diminished levels of haptoglobin, and iron deficiency. Diagnosis is based on the demonstration of the PNH phenotype in a substantial proportion of red cells and granulocytes [[12]].
PNH patients show various degrees of anemia and other bone marrow-related disorders such as granulocytopenia and thrombocytopenia. The incidence of anemia ranges from 88% to 94% and that of leukopenia ranges from 41% to 72%, while that of thrombocytopenia ranges from 51% to 80% [[13],[14]].
Anemia derives from both hemolysis and bone marrow dysfunction that is mostly intravascular [[15]]. Even if hemolysis is continuous during the day, in the night-time, the decline in the blood pH triggers an activation of complement components. This mechanism may be also dependent on an increased nocturnal absorption of lipopolysaccharide (LPS), a major component of the outer membrane of bacteria that stimulates the complement system and is normally bound by monocytes through a GPI-linked protein, CD14, which is lacking in PNH patients [[16], [17],[18]]. The urine is black when the patient awakens, because the amount of free hemoglobin passing through the glomeruli exceeds the absorptive ability of renal tubules [[19]].
Thrombosis represents one of the major determinants of morbidity and mortality in PNH patients [[20]]. In particular, thromboembolism is the principal cause of mortality in this population, leading to 40%–67% of deaths for which the cause is known [[21]]. PNH venous thrombosis often occurs at uncommon sites such as hepatic veins, causing Budd–Chiari syndrome, which represents the principal (40.7%) thrombotic event in these subjects. The second most frequent type of thrombosis is represented by cerebral vein and sinus thrombosis. Pulmonary embolism represents a high-risk site of thrombosis and has been reported in 26 cases, 10 of which were fatal [[22]].
Arterial thrombosis has been described in a minority of subjects, mostly of young age, supporting the hypothesis that it may occur de novo without significant predisposing atherosclerotic disease [[23]].
The pathophysiology of the pro-thrombotic state in PNH is still debated. Ploug et al. [[24]] supposed a failure of the fibrinolytic system, such as a deficiency in urokinase type plasminogen activator receptor on leucocytes. A significant role can be attributed to dysregulated complement activity on GPI anchor-deficient platelets, granulocytes, monocytes and endothelial cells [[25]]. Complement-mediated attack on CD55 and CD59 deficient platelets stimulates the production of factors Va, Xa and prothrombin complex [[26]].
The most-adopted PNH classification was proposed in 2005 by the International PNH Interest Group [[27]], which identified three different kinds of clinical manifestation: classic PNH, which includes patients who have evidence of PNH without bone marrow disorder; PNH associated with another bone marrow disorder [[28]]; and subclinical PNH, in which patients have defective PNH clones without clinical or laboratory signs of hemolysis or thrombosis. De Latour et al. [[29]] added a fourth subgroup defined as intermediate PNH, underlining the fact that many PNH patients do not perfectly fit into these categories because they may suffer from cytopenia or signs of a still-undiagnosed underlying bone marrow failure.
PNH Therapy
In PNH, hemolysis is mostly mediated by the alternative pathway of complement, so complement inhibition therapy is the best strategy against this disease. The choices among the different kinds of complement inhibitor or modulator will be soon enriched by several molecules currently under investigation or recently approved. The activation of complement factor C5, that is at the basis of PNH, generates the potent anaphylatoxin C5a, leading to pathogen lysis, inflammation and cell damage. The first C5 inhibitor was h5G1.1.mAb, later named eculizumab. Eculizumab improved the management of and clinical outcomes in patients with PNH, becoming a proof of concept for other complement-mediated diseases[30].
Eculizumab inhibits the formation of the proinflammatory metabolite C5a and construction of the MAC via C5b, through a mechanism of C5-binding aimed at limiting its cleavage by C5 convertases[307]. Before the approval of eculizumab, the therapeutic alternative for PNH was only supporting the patients by blood transfusion, iron supplementation, anti-thrombosis prophylaxis or therapy and allogeneic bone marrow transplantation[31]. A recent eculizumab-like monoclonal antibody engineered to have a longer half-life is proposed to guarantee the same effects of eculizumab but with a more advantageous and effective dosing schedule: Ravulizumab (ALXN1210)[3230]. This drug represents a new promising instrument for the treatment of PNH, permitting longer dosing intervals of 8 weeks[[3331]]. Despite the increased knowledge of this syndrome, the most appropriate strategy and choice of therapies for PNH are still up for debate. The landscape of combination PNH therapy has markedly progressed over the last few decades, and it will continue to expand with the introduction of long-acting complement inhibitors and the development of complement-modulating drugs. The choice of the right drug is a key component in the improvement of personalized medical care for patients with such a complex disease.
References
- Luzzatto, L.; Paroxysmal nocturnal hemoglobinuria: An acquired X-linked genetic disease with somatic-cell mosaicism.. Curr. Opin. Genet. Dev 2016, 16, 317–322.
- Jennifer J. Johnston; Andrea L. Gropman; Julie C. Sapp; Jamie K. Teer; Jodie M. Martin; Cyndi F. Liu; Xuan Yuan; Zhaohui Ye; Linzhao Cheng; Robert A. Brodsky; et al.Leslie G. Biesecker The Phenotype of a Germline Mutation in PIGA: The Gene Somatically Mutated in Paroxysmal Nocturnal Hemoglobinuria. The American Journal of Human Genetics 2012, 90, 295-300, 10.1016/j.ajhg.2011.11.031.
- Robert A Brodsky; Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Reviews 2008, 22, 65-74, 10.1016/j.blre.2007.10.002.
- T Kinoshita; Dissecting and manipulating the pathway for glycosylphos-phatidylinositol-anchor biosynthesis. Current Opinion in Chemical Biology 2000, 4, 632-638, 10.1016/s1367-5931(00)00151-4.
- Taroh Kinoshita; Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proceedings of the Japan Academy, Series B 2014, 90, 130-143, 10.2183/pjab.90.130.
- Parker, C.J.. Paroxysmal nocturnal hemoglobinuria, Williams Hematology, 9th ed;; McGraw-Hill: New York, NY, USA,, 2015; pp. pp. 571–582..
- Guido Gembillo; Rossella Siligato; Valeria Cernaro; Domenico Santoro; Complement Inhibition Therapy and Dialytic Strategies in Paroxysmal Nocturnal Hemoglobinuria: The Nephrologist’s Opinion. Janus Asbjørn Schatz-Jakobsen; Yuchun Zhang; Krista Johnson; Alyssa Neill; Douglas Sheridan; Gregers Rom Andersen; Structural Basis for Eculizumab-Mediated Inhibition of the Complement Terminal Pathway. The Journal of CliImmunical Medicine ology 2020, 16, 19, 1261, 10.3390/jcm9051261.7, 337-344, 10.4049/jimmunol.1600280.
- Guido Gembillo; Rossella Siligato; Valeria Cernaro; Domenico Santoro; Complement Inhibition Therapy and Dialytic Strategies in Paroxysmal Nocturnal Hemoglobinuria: The Nephrologist’s Opinion. JoAnita Hill; Amy E. DeZern; Taroh Kinoshita; Robert A. Brodsky; Paroxysmal nocturnal haemoglobinuria. Naturnale of Clinical MedicinReviews Disease Prime rs 2020, 9, 1261, 10.3390/jcm9051261.17, 3, 17028, 10.1038/nrdp.2017.28.
- Anita Hill; Amy E. DeZern; Taroh Kinoshita; Robert A. Brodsky; Paroxysmal nocturnal haemoglobinuria. NatC Armstrong; J Schubert; E Ueda; J J Knez; D Gelperin; S Hirose; R Silber; S Hollan; R E Schmidt; M E Medof; et al. Affected paroxysmal nocturnal hemoglobinuria T lymphocytes harbor a common defect in assembly of N-acetyl-D-glucosamine inositol phospholipid corresponding to that in class A Thy-1- murine lymphoma mutants.. Jourenal of Reviews Disease Primers Biological Chemistry 1992017, 3, 17028, 10.1038/nrdp.2017.28., 267, 25347-51..
- C Armstrong; J Schubert; E Ueda; J J Knez; D Gelperin; S Hirose; R Silber; S Hollan; R E Schmidt; M E Medof; et al. Affected paroxysmal nocturnal hemoglobinuria T lymphocytes harbor a common defect in assembly of N-acetyl-D-glucosamine inositol phospholipid corresponding to that in class A Thy-1- murine lymphoma mutants.. JM. Hidaka; S. Nagakura; K. Horikawa; T. Kawaguchi; N. Iwamoto; T. Kagimoto; K. Takatsuki; H. Nakakuma; Impaired Glycosylation of Glycosylphosphatidylinositol-Anchor Synthesis in Paroxysmal Nocturnal Hemoglobinuria Leukocytes. Biournchemical of Biological Chemiand Biophysical Research Communicationstry 1992, 267, 25347-51..3, 191, 571-579, 10.1006/bbrc.1993.1256.
- M. Hidaka; S. Nagakura; K. Horikawa; T. Kawaguchi; N. Iwamoto; T. Kagimoto; K. Takatsuki; H. Nakakuma; Impaired Glycosylation of Glycosylphosphatidylinositol-Anchor Synthesis in Paroxysmal Nocturnal Hemoglobinuria Leukocytes. Biochemical and Biophysical Research Communications 1993, 191, 571-579, 10.1006/bbrc.1993.1256.Keohane, E.M., Otto, C.N., Walenga, J.M.,. Intrinsic defects leading to increased erythrocyte destruction. Rodak’s Hematology, 6th ed; Elsevier: Amsterdam, 2020; pp. pp. 336–362..
- Keohane, E.M., Otto, C.N., Walenga, J.M.,. Intrinsic defects leading to increased erythrocyte destruction. Rodak’s Hematology, 6th ed; Elsevier: Amsterdam, 2020; pp. pp. 336–362..Lucio Luzzatto; Giacomo Gianfaldoni; Recent Advances in Biological and Clinical Aspects of Paroxysmal Nocturnal Hemoglobinuria. International Journal of Hematology 2006, 84, 104-112, 10.1532/ijh97.06117.
- Lucio Luzzatto; Giacomo Gianfaldoni; Recent Advances in Biological and Clinical Aspects of Paroxysmal Nocturnal Hemoglobinuria. IntG. Sociè; J Y Mary; A De Gramont; B Rio; M Leporrier; C Rose; P Heudier; H Rochant; J Y Cahn; Eliane Gluckman; et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology.. Thern Lational Journal of Hemancetology 200 1996, , 3484, 104-112, 10.1532/ijh97.06117., 573-577.
- G. Sociè; J Y Mary; A De Gramont; B Rio; M Leporrier; C Rose; P Heudier; H Rochant; J Y Cahn; Eliane Gluckman; et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology.. ThJun-Ichi Nishimura; Yuzuru Kanakura; Russell E. Ware; Tsutomu Shichishima; Hideki Nakakuma; Haruhiko Ninomiya; Carlos M. DeCastro; Sharon Hall; Akihisa Kanamaru; Keith M. Sullivan; et al.Hideaki MizoguchiMitsuhiro OmineTaroh KinoshitaWendell F. Rosse Clinical Course and Flow Cytometric Analysis of Paroxysmal Nocturnal Hemoglobinuria in the United States and Japan. Me Ladicincet 1996, 2004, 8348, 573-577., 193-207, 10.1097/01.md.0000126763.68170.46.
- Jun-Ichi Nishimura; Yuzuru Kanakura; Russell E. Ware; Tsutomu Shichishima; Hideki Nakakuma; Haruhiko Ninomiya; Carlos M. DeCastro; Sharon Hall; Akihisa Kanamaru; Keith M. Sullivan; et al.Hideaki MizoguchiMitsuhiro OmineTaroh KinoshitaWendell F. Rosse Clinical Course and Flow Cytometric Analysis of Paroxysmal Nocturnal Hemoglobinuria in the United States and Japan. MW F Rosse; Evolution of clinical understanding: paroxysmal nocturnal hemoglobinuria as a paradigm.. Amedricinan Journal of He 2004, 83, 193-207, 10.1097/01.md.0000126763.68170.46.matology 1993, 42, 122-126.
- W F Rosse; Evolution of clinical understanding: paroxysmal nocturnal hemoglobinuria as a paradigm.. AmC Couturier; Nicole Haeffner-Cavaillon; M Caroff; M D Kazatchkine; Binding sites for endotoxins (lipopolysaccharides) on human monocytes.. Therican Journal of HematImmunology 1993, 1, 142, 122-126.7, 1899-1904.
- C Couturier; Nicole Haeffner-Cavaillon; M Caroff; M D Kazatchkine; Binding sites for endotoxins (lipopolysaccharides) on human monocytes.. The JournaDl Simmons; S Tan; Dg Tenen; A Nicholson-Weller; B Seed; Monocyte antigen CD14 is a phospholipid anchored membrane protein. Bl of Immunology od 19891, 14, 7, 1899-1904.3, 284-289, 10.1182/blood.v73.1.284.284.
- Dl Simmons; S Tan; Dg Tenen; A Nicholson-Weller; B Seed; Monocyte antigen CD14 is a phospholipid anchored membrane protein. BSaleh Rachidi; Khaled M. Musallam; Ali Taher; A closer look at paroxysmal nocturnal hemoglobinuria. European Journal oof Internal Med icine 201989, 73, 284-289, 10.1182/blood.v73.1.284.284.0, 21, 260-267, 10.1016/j.ejim.2010.04.002.
- Saleh Rachidi; Khaled M. Musallam; Ali Taher; A closer look at paroxysmal nocturnal hemoglobinuria. Robert S. Schwartz; Black Mornings, Yellow Sunsets — A Day with Paroxysmal Nocturnal Hemoglobinuria. New Europengland Journal of Internal Medicine 2010, 21, 260-267, 10.1016/j.ejim.2010.04.002.4, 350, 537-538, 10.1056/nejmp038223.
- Robert S. Schwartz; Black Mornings, Yellow Sunsets — A Day with Paroxysmal Nocturnal Hemoglobinuria. New England S. T. A. Van Bijnen; W. L. Van Heerde; Petra Muus; Mechanisms and clinical implications of thrombosis in paroxysmal nocturnal hemoglobinuria. Journal of MedThrombosicins and Hae mostasis 2004, 3512, 10, 537-538, 10.1056/nejmp038223., 1-10, 10.1111/j.1538-7836.2011.04562.x.
- Tomosato Yamazaki; Kensuke Suzuki; Masaaki Sumi; Kiyoyuki Yanaka; Hiroshi Kojima; Akira Matsumura; Cerebral Embolism as a Complication of Paroxysmal Nocturnal Hemoglobinuria. EurAnita Hill; Richard J. Kelly; Peter Hillmen; Thrombosis in paroxysmal nocturnal hemoglobinuria. Blopean Neurology d 2005, 53, 217-220, 10.1159/000086735.13, 121, 4985-4996, 10.1182/blood-2012-09-311381.
- M Ploug; T Plesner; E Ronne; V Ellis; G Hoyer-Hansen; Ne Hansen; K Dano; The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. BP. D. Ziakas; L. S. Poulou; G. I. Rokas; D. Bartzoudis; M. Voulgarelis; Thrombosis in paroxysmal nocturnal hemoglobinuria: sites, risks, outcome. An overview. Journal oof Thrombosis and 199 Haemostasis 2, 79, 1447-1455, 10.1182/blood.v79.6.1447.bloodjournal7961447.007, 5, 642-645, 10.1111/j.1538-7836.2007.02379.x.
- Ilene Ceil Weitz; Thrombosis in Patients with Paroxysmal Nocturnal Hemoglobinuria. STomosato Yamazaki; Kensuke Suzuki; Masaaki Sumi; Kiyoyuki Yanaka; Hiroshi Kojima; Akira Matsumura; Cerebral Embolism as a Complication of Paroxysmal Nocturnal Hemoglobinuria. Europeminars in Thrombosis and Hemostasis n Neurology 2011, 05, 537, 315-321, 10.1055/s-0031-1273095., 217-220, 10.1159/000086735.
- Panayiotis D Ziakas; Loukia S Poulou; Anastasia Pomoni; Thrombosis in paroxysmal nocturnal hemoglobinuria at a glance: a clinical review.. Current VascuM Ploug; T Plesner; E Ronne; V Ellis; G Hoyer-Hansen; Ne Hansen; K Dano; The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blar Pharmacology od 1992008, 6, 347-353, 10.2174/157016108785909742., 79, 1447-1455, 10.1182/blood.v79.6.1447.bloodjournal7961447.
- Charles Parker; Mitsuhiro Omine; Stephen J. Richards; Jun-Ichi Nishimura; Monica Bessler; Russell Ware; Peter Hillmen; Lucio Luzzatto; Neal Young; Taroh Kinoshita; et al.Wendell RosseGérard SociéInternational PNH Interest Group Diagnosis and management of paroxysmal nocturnal hemoglobinuria.. BlIlene Ceil Weitz; Thrombosis in Patients with Paroxysmal Nocturnal Hemoglobinuria. Seminars in Thrombosis and Hemostasis 2005, 106, 3699-709, 10.1182/blood-2005-04-1717.11, 37, 315-321, 10.1055/s-0031-1273095.
- Charles Parker; Mitsuhiro Omine; Stephen J. Richards; Jun-Ichi Nishimura; Monica Bessler; Russell Ware; Peter Hillmen; Lucio Luzzatto; Neal Young; Taroh Kinoshita; et al.Wendell RosseGérard SociéInternational PNH Interest Group Diagnosis and management of paroxysmal nocturnal hemoglobinuria.. BPanayiotis D Ziakas; Loukia S Poulou; Anastasia Pomoni; Thrombosis in paroxysmal nocturnal hemoglobinuria at a glance: a clinical review.. Current Vascular Pharmacood logy 2005, 108, 6, 3699-709, 10.1182/blood-2005-04-1717., 347-353, 10.2174/157016108785909742.
- Brodsky, R.A.. Paroxysmal nocturnal hemoglobinuria; Hematology: Basic Principles and Practice, 7th ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. pp. 415–424..Charles Parker; Mitsuhiro Omine; Stephen J. Richards; Jun-Ichi Nishimura; Monica Bessler; Russell Ware; Peter Hillmen; Lucio Luzzatto; Neal Young; Taroh Kinoshita; et al.Wendell RosseGérard SociéInternational PNH Interest Group Diagnosis and management of paroxysmal nocturnal hemoglobinuria.. Blood 2005, 106, 3699-709, 10.1182/blood-2005-04-1717.
- Régis Peffault De Latour; Jean Yves Mary; Célia Salanoubat; Louis Terriou; Gabriel Etienne; Mohamad Mohty; Sophie Roth; Sophie De Guibert; Sebastien Maury; Jean Yves Cahn; et al.Gérard Sociéon behalf of the French Society of Hematology and of the French Association of Young Hematologists Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 2008, 112, 3099-3106, 10.1182/blood-2008-01-133918.Brodsky, R.A. Hematology: Basic Principles and Practice, 7th ed.; Paroxysmal nocturnal hemoglobinuria; Elsevier: Philadelphia, PA, USA, 2018; pp. 415–424
- Régis Peffault De Latour; Jean Yves Mary; Célia Salanoubat; Louis Terriou; Gabriel Etienne; Mohamad Mohty; Sophie Roth; Sophie De Guibert; Sebastien Maury; Jean Yves Cahn; et al.Gérard Sociéon behalf of the French Society of Hematology and of the French Association of Young Hematologists Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 2008, 112, 3099-3106, 10.1182/blood-2008-01-133918.
- Janus Asbjørn Schatz-Jakobsen; Yuchun Zhang; Krista Johnson; Alyssa Neill; Douglas Sheridan; Gregers Rom Andersen; Structural Basis for Eculizumab-Mediated Inhibition of the Complement Terminal Pathway. Robert M. Stern; Nathan T. Connell; Ravulizumab: a novel C5 inhibitor for the treatment of paroxysmal nocturnal hemoglobinuria. Therapeutic Journal of ImmunAdvances in Hematology 2016, 9, 197, 337-344, 10.4049/jimmunol.1600280.0, null, 10.1177/2040620719874728.
- Lucio Luzzatto; Recent advances in the pathogenesis and treatment of paroxysmal nocturnal hemoglobinuria. F1000RAlexander Röth; Scott T. Rottinghaus; Anita Hill; Eric S. Bachman; Jin Seok Kim; Hubert Schrezenmeier; Louis Terriou; Álvaro Urbano-Ispizua; Richard A. Wells; Jun Ho Jang; et al.Austin G. KulasekararajJeff SzerRasha AguzziAndrew I. DamokoshLori ShafnerJong Wook Lee Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Advancesearch 2016, 5, 209, 10.12688/f1000research.7288.1.8, 2, 2176-2185, 10.1182/bloodadvances.2018020644.
- European Medicines Agency. Ultomiris, Authorisation Details. Available online: . www.ema.europa.eu. Retrieved 2020-5-3
- Alexander Röth; Scott T. Rottinghaus; Anita Hill; Eric S. Bachman; Jin Seok Kim; Hubert Schrezenmeier; Louis Terriou; Álvaro Urbano-Ispizua; Richard A. Wells; Jun Ho Jang; et al.Austin G. KulasekararajJeff SzerRasha AguzziAndrew I. DamokoshLori ShafnerJong Wook Lee Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Advances 2018, 2, 2176-2185, 10.1182/bloodadvances.2018020644.
- Alexander Röth; Scott T. Rottinghaus; Anita Hill; Eric S. Bachman; Jin Seok Kim; Hubert Schrezenmeier; Louis Terriou; Álvaro Urbano-Ispizua; Richard A. Wells; Jun Ho Jang; et al.Austin G. KulasekararajJeff SzerRasha AguzziAndrew I. DamokoshLori ShafnerJong Wook Lee Ravulizumab (ALXN1210) in patients with paroxysmal nocturnal hemoglobinuria: results of 2 phase 1b/2 studies. Blood Advances 2018, 2, 2176-2185, 10.1182/bloodadvances.2018020644.