Aging can be seen as process characterized by accumulation of oxidative stress induced damage. Oxidative stress derives from different endogenous and exogenous processes, all of which ultimately lead to progressive loss in tissue and organ structure and functions. The oxidative stress theory of aging expresses itself in age-related diseases. Aging is in fact a primary risk factor for many diseases and in particular for cardiovascular diseases and its derived morbidity and mortality. Here we highlight the role of oxidative stress in age-related cardiovascular aging and diseases. We take into consideration the molecular mechanisms, the structural and functional alterations, and the diseases accompanied to the cardiovascular aging process.
https://www.mdpi.com/2075-1729/11/1/60
- aging
- oxidative stress
- cardiovascular
1. Behind the scenes of Cardiovascular Aging
1.1. Aging, Oxidative Stress and Cellular Dysfunction
Age-dependent oxidative stress cellular dysfunction is a condition determined by various intrinsic factors. The heart cell, the cardiomyocytes, exert a high aerobic metabolism in order to guarantee their functions. In this scenario cellular metabolism is guaranteed by mitochondrial function [1][1]. Mitochondria occupy 30% of total cardiomyocyte volume, this guarantees the high energy demands of the heart. Mitochondrial oxidative phosphorylation fueled by catabolism of lipids and carbohydrates is the main source of ATP for heart function. The Krebs cycle is important for ATP generation but its balance and regulation are fundamental for oxidative stress management [2][2]. Cardiomyocytes aging is one of the main factors leading to cardiovascular diseases. Understanding the processes behind cellular dysfunction in the aging process has led to many findings in relation to mitochondrial dysfunction and its genomic instability and epigenetic regulation [3][3]. In fact, studies on longevity have highlighted the importance of mitochondria and oxidative stress in the senescence process [4][4]. As described before, free radicals are generated in different compartments of the cell. However, the main source of free radicals is mitochondria and in particular the electron transport chain located in the inner mitochondrial membrane. The continuous production of free radicals in the mitochondria leads to oxidation of macromolecules and DNA [5][5]. In particular, during aging nuclear and mitochondrial DNA stability is compromised [6][6]. Mitochondrial DNA lesions have been shown to carry mutagenic and cytotoxic affects ultimately leading to DNA replication alterations [7,8][7][8]. The genomic instability is partially prevented by DNA repair pathways in both mitochondria and nucleus [9][9]. Despite this DNA damage increases and accumulates during the aging process. Aged mice heart mitochondria have a threefold higher frequency of mitochondrial DNA mutation and deletions [10][10]. Accumulation of DNA alterations is a driving factor in aging leading to cellular loss of function. Animal models with increased mitochondrial DNA instability mediated by disruption of mitochondrial transcription factor A, showed alteration of mitochondrial morphology leading to higher rates of apoptosis and ultimately to dilated cardiomyopathy [11, 12][11][12]. An early aging phenotype is characterized by high mitochondrial DNA point mutation as highlighted in mice [13,14][13][14]. New animal models have been developed to study the effect of increased mitochondrial DNA deletions in the myocardium. These models have highlighted that mitochondrial DNA dysfunction is not only associated with tissue dysfunction but also with development of premature arrhythmias [15][15]. Mitochondrial dysfunction and instability contribute to the aging process in other ways as is the case with calcium homeostasis and redox signaling [16,17][16][17]. Overall, the aging process and the mitochondrial alterations lead to metabolic changes. Normally up to 90% of cardiac ATP is generated by the use of long-chain fatty acid, only 10% from glucose, lactate, ketone bodies, and amino acids [18,19][18][19]. The Randle cycle highlights the substrate selection and interaction. Development of heart disease and aging has been associated with the inversion of metabolic substrate selection [20][20]. With aging increased glucose metabolism expression, increased glycolytic proteins and the concomitant decline in fatty acid oxidation has shown to be a premature sign of heart failure in younger individuals [21][21]. As we will mention afterwards, metabolic inversion and heart failure are associated with a reactive hyper-adrenergic state closely linked to impaired glucose metabolism and inevitably to insulin resistance [22][22].
Another important cellular dysfunction point to address is increased apoptosis during aging [23][23]. Accumulation of lipofuscin on lysosomes, loss of autophagic management, accumulation of non-functional damaged cell components, in particular mitochondria, leads to activation of the apoptotic pathway [24][24]. These changes in cardiomyocytes also increase alteration in calcium cellular handling. Calcium plays a crucial role in modulating cardiac function by modulation of myocardial contractility through a complex system of ion channels, ryanodine receptors, and sodium calcium exchangers. On the other hand, myocardial relaxation is mediated by sarcoplasmatic/endoplasmatic reticulum calcium-ATPase (SERCA2a) which seizes calcium to the sarcoplasmic reticulum [25][25]. The importance of cellular SERCA2a function has been highlighted in oxidative stress and aging. In particular, SERCA2a impairment and consequent increase in calcium stagnation in cytoplasm causes diastolic relaxation impairment. Moreover, animal models by the same mechanism of prolonged cellular contraction showed alterations in myosin heavy chain expression [25][25]. These changes influence negatively cardiomyocytes contractile and electrical efficiency [66]. Alteration of the contraction and relaxation coupling of cardiomyocytes is at the base of the compensation mechanism that we will describe in the next chapters. In fact, we will see how depletion of cardiomyocytes and inadequate contraction efficiency leads to cardiac hypertrophic remodeling and not only [26][26].
1.2. Aging in Cardiac Structural and Patho-Physiological Changes
Cardiac structural modifications due to aging bring about various functional and adaptive consequences in heart capacity [27][27]. Cardiac aging causes fibrosis, left ventricle hypertrophy, diastolic dysfunction, and filling ultimately leading to reduction in cardiac overall compliance and ejection fraction output [28, 29][28][29]. Gene expression in aging human hearts highlights a shift in cellular capacity. Upregulation of genes for sarcomere and cytoskeletal proteins and downregulation of proteasome components expression reflects the process of myocyte hypertrophy and decreased in protein turnover [30][30]. This is accompanied by downregulation in Troponin T, SERCA2, and alpha-MHC with a switch towards beta-MHC, with a decrement in contractile efficiency [31][31]. Mitochondrial disfunction in mice aging cardiomyocytes is an important source of oxidative stress [32][32]. The inefficiency of aging rat mitochondria is characterized by increase in size, reduction in number of cristae with an overall reduction in ATP production per cell [33][33]. The high-rate production of reactive oxygen species leads to a vicious cycle [34,35][34][35]. In fact, in order to maintain functional demands the aging heart undergoes a process of hypertrophy [36][36]. This adaptation allows the heart to respond to increase in pressure demand in consequence to vascular stiffness [37][37]. However, this mechanism fails when heart demands exceed bringing about heart failure [38][38]. Cardiac hypertrophy in human aging-heart is characterized by increase in cardiomyocyte size with asymmetric left ventricle ventricular wall thickening mainly of the intraventricular septum [39][39]. These alterations cause alterations in human heart geometry and shape initially granting increase in contractility but on the long run causing oxygen impairment and reduction in contractility [40][40]. Aging in both mice models and humans is associated with apoptosis and loss of myocytes with a rate of about 45 million per year compensated by increase in single myocyte median volume [41,42][41][42]. The overload of the remaining myocytes is an additional reason for compensatory hypertrophy [43][43]. In addition, the loss of vascular elasticity and the increase in vascular stiffness increases the mechanical load of the aging heart accelerating the way to heart failure [44][44]. Age-related left ventricle hypertrophy in humans occurs independently of underlining story of hypertension or other causes [45][45]. Overall, a healthy human aging heart with preserved ejection fraction (EF) shows no change in stroke volume, heart rate, and cardiac output [46][46]. The main difference is highlight in diastolic function, in fact, the capacity of the ventricle to receive blood results altered by the reduction in ventricle wall elastic compliance [47][47]. Hemodynamically this can be seen in a reduction in early diastolic filling with an increase in tele systolic filling with an inversion of the E/A parameter and an increase in E/e’ better seen in echocardiographic studies [48,49][48][49]. This compensatory system present at rest however is not efficacious in case of cardiac demands. The aging-heart in fact compensates the contractile and diastolic function reduction with increase in peak heart rate [50][50]. Another important age-related alteration in animal model hearts is the calcification of the valves associated with proliferation of fibroblasts and deposition of collagen [51,52][51][52]. The cardiac extracellular matrix in the aging heart fills with glycoproteins, proteoglycans, glycosaminoglycans, integrins, and collagen [53-54][53][54]. Interesting is the switch from collagen type III to type I, seen in children, young adults, and elderly, which is less distensible and stiffer [55,56][55][56]. This also effects negatively the propagation of the electrical signals of the myocytes and of the myofibrillar bundles giving arrhythmias [57,58][57][58].
1.3. Aging and Autonomic Nervous System Adaptations
With aging an increasing plasma catecholamine concentrations and sympathetic tone of autonomic nervous system (ANS) [59,60][59][60] could impair adaptivity of elderly individuals to the environment [61][61]. Aging has been implicated in the impairment of alfa and beta-adrenergic receptor sensitivity and vascularresponsiveness[62][63] [62,63] with a predominance of the alfa tone in muscle vasculature. In fact, in older women we can observe a higher decrease of arterial blood pressure in front of younger ones after an autonomic blockade [64][64]. Age-related alterations in ANS influence blood pressure, cerebral blood flow, bladder function, and heart rate variability (HRV) [65,66][65][66]. HRV is an indicator of arrhythmic complications and a strong predictor of mortality and sudden death [67][67]. Nocturnal reduction of parasympathetic activity in elderly individuals is the result of a low vagal hearth control due to the prevalence of the adrenergic tone of SNA. The increase in heart rate combined with the HRV reduction is due to the degeneration of cardiac autonomic function during aging[68] [68] and increases the incidence of cardiovascular events[69] [69]. Active lifestyle is important for the health of the elderly. Intensive physical exercise, increasing muscle mass and fatigue resistance, could minimize autonomic dysfunction in aging with training [70][70]. A physical training regime could improve adaptations of the autonomic function [71][71]. Although intensive exercise seems to have benefit for older individuals, there is a need for further studies to realize a regimen of exercise programs. The ANS abnormalities were thought to be a common underlying pathophysiology of CVDs such as hypertension and heart failure [72][72]. Another issue due to the impairment between the two ANS deregulation is atrial fibrillation, the most common arrhythmia in older people [73][73]. Denervation of ANS has been shown to be efficient against AF [74][74]. Patients with AF had reduction in cardiac performance due to the loss of the atrial contraction in ventricular filling.
3. Aging, Oxidative Stress, and Cardiovascular Diseases
2. Aging, Oxidative Stress, and Cardiovascular Diseases
As highlighted above, all cells constantly undergo reactions which require transfer of electrons, in order to acquire energy
As highlighted above, all cells constantly undergo reactions which require transfer of electrons, in order to acquire energy . These complex mechanisms, which occur principally in the mitochondria, require a constant turnover in the oxidative state[75]. The redox reactions and the reactive oxygen species (ROS) generated are highly reactive and are the main source of oxidative stress. The heart’s cellular volume is made up for 45% by mitochondria
. These complex mechanisms, which occur principally in the mitochondria, require a constant turnover in the oxidative state [75]. The redox reactions and the reactive oxygen species (ROS) generated are highly reactive and are the main source of oxidative stress. The heart’s cellular volume is made up for 45% by mitochondria [181]. Oxidative phosphorylation generates radical species, when electrons are lost from mitochondrial complexes I and II, in order to produce adenosine triphosphate (ATP) [76][76]. The mitochondrial pathway also characterized by formation of NADH and FADH which react with other redox compounds [77][77]. As cardiomyocytes and endothelial cells age, they produce more ROS. Increased production of ROS leads to functional impairment due to reduction in biological activity of nitric oxide and formation of peroxynitrite, which deactivates several free radical scavengers [78-79][78][79]. Another important origin of oxidative stress is aberrant Ca2+ reuptake, due to SERCA2 downregulation [80-81][80][81]. Constant high concentration of Ca2+ levels in the cardiomyocyte’s cytosol increases ROS production [184,185]. As highlighted before the RAAS system also plays an important role in age-related cardiac oxidative stress with direct and indirect involvement of NADPH oxidase complex and of ROS production [82][82]. Thus age-dependent increase in oxidative damage is the one of the leading causes of CVD (Figure 1) [83][83].
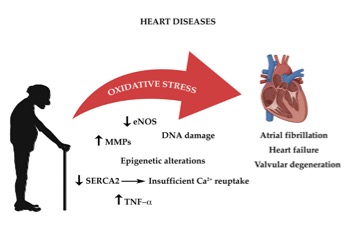
Figure 1. The impact of oxidative stress on age-associated heart diseases
3.1. Atrial Fibrillation
Atrial fibrillation is the most common chronic cardiac rhythm disturbance, it affects 1–2% of the population and the chances of developing this condition increases with age, particularly after age 65 [84]. It consists in a rapid heartbeat (tachyarrhythmia) that originates in the upper chambers of the heart, called the atria, preventing them from functioning properly [85]. In such circumstances, the atria are no longer able to expel all the blood, which will remain partially inside the atria with the risk of clot formation [86]. Clot formation due to AF is one the leading causes of cerebrovascular thrombosis [87,88]. In aging the main cause of AF can be found in the hearts structural and functional alteration, as described earlier. Aging itself is the single most important risk factor for AF [89]. Among all the previously described alteration, the electrical conduction disturbances, ectopic activity leading to atrial arrhythmogenesis is atrial fibrosis and so dilation [90]. Atrial fibrosis in the human aging heart is associated with excessive accumulation of collagen fibers, in particular type I fibers, and the increase of cross-linking between fibers [91]. Oxidative stress plays an important role in extracellular matrix (ECM) turnover and metabolism. The increase of matrix metalloproteinase (MMP) in human aging hearts with atrial fibrillation has been widely highlighted [92]. Age-related alteration are a summation of structural and electrophysiological remodeling leading to alteration of the mechano-electrical feedback [93]. Overall, this means that the atria are no longer capable of modulating the electric functions of the heart with the induction of mechanical load on the cardiomyocytes [94]. In conclusion, aging is associated with an increase in acute-phase inflammatory cytokines an important stimulus for AF, as it directly increases arrhythmogenicity and calcium homeostasis dysregulation [95,96]. Moreover, as seen in human atrial tissues, ROS and mitochondrial disfunction influence AF due to accumulation of age-related mitochondrial DNA deletion and mutation [97,98]. Oxidative stress and inflammation in aging influence greatly both functional and structural modifications causing electrophysiological remodeling and correlated diseases [99,100].
Figure 1.The impact of oxidative stress on age-associated heart diseases
2.1. Atrial Fibrillation
3
Atrial fibrillation is the most common chronic cardiac rhythm disturbance, it affects 1–2% of the population and the chances of developing this condition increases with age, particularly after age 65[84]. It consists in a rapid heartbeat (tachyarrhythmia) that originates in the upper chambers of the heart, called the atria, preventing them from functioning properly85]. In such circumstances, the atria are no longer able to expel all the blood, which will remain partially inside the atria with the risk of clot formation][86]. Clot formation due to AF is one the leading causes of cerebrovascular thrombosis[87][88]. In aging the main cause of AF can be found in the hearts structural and functional alteration, as described earlier. Aging itself is the single most important risk factor for AF[89] Among all the previously described alteration, the electrical conduction disturbances, ectopic activity leading to atrial arrhythmogenesis is atrial fibrosis and so dilation[90]. Atrial fibrosis in the human aging heart is associated with excessive accumulation of collagen fibers, in particular type I fibers, and the increase of cross-linking between fibers[91]. Oxidative stress plays an important role in extracellular matrix (ECM) turnover and metabolism. The increase of matrix metalloproteinase (MMP) in human aging hearts with atrial fibrillation has been widely highlighted[92]. Age-related alteration are a summation of structural and electrophysiological remodeling leading to alteration of the mechano-electrical feedback[93]. Overall, this means that the atria are no longer capable of modulating the electric functions of the heart with the induction of mechanical load on the cardiomyocytes[94]. In conclusion, aging is associated with an increase in acute-phase inflammatory cytokines an important stimulus for AF, as it directly increases arrhythmogenicity and calcium homeostasis dysregulation[95][96]. Moreover, as seen in human atrial tissues, ROS and mitochondrial disfunction influence AF due to accumulation of age-related mitochondrial DNA deletion and mutation[97][98]. Oxidative stress and inflammation in aging influence greatly both functional and structural modifications causing electrophysiological remodeling and correlated diseases[99][100].2. Heart Failure
2.2. Heart Failure
Heart failure (HF) is a complex clinical syndrome defined by the inability of the heart to supply blood in adequate quantities for the body’s actual demand or the inability to meet this demand only at ventricular filling pressures above the norm [101]
Heart failure (HF) is a complex clinical syndrome defined by the inability of the heart to supply blood in adequate quantities for the body’s actual demand or the inability to meet this demand only at ventricular filling pressures above the norm[101]. According to statistics, as the population ages and the number of patients who survive a myocardial infarction increases, the incidence of heart failure continues to rise[102]. If we refer to hemodynamics, heart failure is characterized by reduced contractility of the myocardium measured as ejection fraction (EF), this universally used parameter can actually be not very specific in identifying the cause of cardiac dysfunction. In fact, this condition can be caused by both organic and functional problems. Among the most common causes are myocardial infarction, myocardial ischemia, hypertension, valvulopathies, cardiomyopathies, metabolic diseases, and autoimmune diseases
. According to statistics, as the population ages and the number of patients who survive a myocardial infarction increases, the incidence of heart failure continues to rise [102]. If we refer to hemodynamics, heart failure is characterized by reduced contractility of the myocardium measured as ejection fraction (EF), this universally used parameter can actually be not very specific in identifying the cause of cardiac dysfunction. In fact, this condition can be caused by both organic and functional problems. Among the most common causes are myocardial infarction, myocardial ischemia, hypertension, valvulopathies, cardiomyopathies, metabolic diseases, and autoimmune diseases. HF is the most important complication of any heart disease. In the United States of America, it is estimated that in 2006 there were more than 600,000 new cases [103][103]. In Italy about 5% of the general population (3,000,000 individuals) is affected by overt or asymptomatic HF [104][104]. Age is a very important risk factor, the incidence remains low in people between 40 and 50 years, while it rises up to 10% in people over 75 years of age [105][105]. The incidence of this pathology is in rapid increase due to the overall increased survival rate following acute myocardial infarction and to longer life expectancy [106][106]. Aging (with consequent organ degeneration) causes cardiac efficiency to decrease, amplifying the effect of any pathologies. HF is generally thought to be involved in all those deaths from aging without apparent symptoms. In fact, when cardiac contractility is progressively reduced, whatever the cause, the final effect is hypo-perfusion of the organs vital and in particular of the brain, kidneys, and liver with the progressive functional deterioration [107][107].
Systolic HF is characterized by the reduced performance of the left ventricle, easily identifiable in patients with a universally adopted echocardiographic parameter such as the ejection fraction (FE), which, however, can vary considerably with the different biomedical imaging methods[108]. The force of contraction of the heart is directly proportional to the conditions of the myocyte, the cell of which the heart muscle is composed: any insult that affects the myocytes is reflected on the compliance of the left ventricle and therefore on the force of contraction (the FE is generally lower than 45%)[109]. Ventricular hypertrophy is one of the adaptation mechanisms of the heart subjected to increased stress that persists for long periods of time; this attempt at correction may be a factor involved in the progression of heart failure[110]. It is certainly not the mechanism that leads to myocardial hypertrophy, it is certain that an increase in systolic wall tension in association with an increase in afterload would cause concentric hypertrophy; on the contrary, an increase in diastolic wall tension in association with an increase in preload would lead to eccentric hypertrophy[111]. In both situations the synthesis of the unit of the myocyte known as sarcomere would be stimulated: in the first case the production of sarcomeres would be stimulated in parallel and in the second in series[111]. Myocytes and their components can be damaged by inflammatory diseases (myocarditis) and by infiltrates (amyloidosis), by toxins or by drugs. The most common mechanism is certainly myocardial ischemia: with the death of myocytes, the myocardium is replaced with fibrous or connective tissue, which have no contractile properties and are similar to scars. These scars can initiate the heart remodeling process, which in turn can lead to heart failure[113]. Heart failure caused by diastolic dysfunction, like systolic dysfunction, may be symptom-free in a compensated patient[114]. What characterizes this alteration is the inability of the left ventricle to adequately relax and this is secondary to the increased stiffness of the ventricular chamber. This attitude of the heart muscle leads to a reduced ventricular filling in diastole, which translates into a reduction in output. The inability to obtain optimal relaxation leads to an increase in end-diastolic pressures, which affect the atria and pulmonary veins[115]. Diastolic dysfunction and systolic dysfunction have many causes in common, most notably older age, high blood pressure, diabetes mellitus, and left ventricular hypertrophy. We can consider separately female sex, diseases of the pericardium and hypertrophic, accumulation and infiltrative cardiomyopathies[116]. Restrictive cardiomyopathy is one of the diseases that most affect diastolic dysfunction. Restrictive cardiomyopathies are characterized by a restrictive filling and a reduced diastolic volume; they are classified into primary and secondary[117].
Systolic HF is characterized by the reduced performance of the left ventricle, easily identifiable in patients with a universally adopted echocardiographic parameter such as the ejection fraction (FE), which, however, can vary considerably with the different biomedical imaging methods [108]. The force of contraction of the heart is directly proportional to the conditions of the myocyte, the cell of which the heart muscle is composed: any insult that affects the myocytes is reflected on the compliance of the left ventricle and therefore on the force of contraction (the FE is generally lower than 45%) [109]. Ventricular hypertrophy is one of the adaptation mechanisms of the heart subjected to increased stress that persists for long periods of time; this attempt at correction may be a factor involved in the progression of heart failure [110]. It is certainly not the mechanism that leads to myocardial hypertrophy, it is certain that an increase in systolic wall tension in association with an increase in afterload would cause concentric hypertrophy; on the contrary, an increase in diastolic wall tension in association with an increase in preload would lead to eccentric hypertrophy [111]. In both situations the synthesis of the unit of the myocyte known as sarcomere would be stimulated: in the first case the production of sarcomeres would be stimulated in parallel and in the second in series [112]. Myocytes and their components can be damaged by inflammatory diseases (myocarditis) and by infiltrates (amyloidosis), by toxins or by drugs. The most common mechanism is certainly myocardial ischemia: with the death of myocytes, the myocardium is replaced with fibrous or connective tissue, which have no contractile properties and are similar to scars. These scars can initiate the heart remodeling process, which in turn can lead to heart failure [113]. Heart failure caused by diastolic dysfunction, like systolic dysfunction, may be symptom-free in a compensated patient [114]. What characterizes this alteration is the inability of the left ventricle to adequately relax and this is secondary to the increased stiffness of the ventricular chamber. This attitude of the heart muscle leads to a reduced ventricular filling in diastole, which translates into a reduction in output. The inability to obtain optimal relaxation leads to an increase in end-diastolic pressures, which affect the atria and pulmonary veins [115]. Diastolic dysfunction and systolic dysfunction have many causes in common, most notably older age, high blood pressure, diabetes mellitus, and left ventricular hypertrophy. We can consider separately female sex, diseases of the pericardium and hypertrophic, accumulation and infiltrative cardiomyopathies [116]. Restrictive cardiomyopathy is one of the diseases that most affect diastolic dysfunction. Restrictive cardiomyopathies are characterized by a restrictive filling and a reduced diastolic volume; they are classified into primary and secondary [117].
In the failing aging heart, oxidative stress plays an important role. Both normal aging and pathological aging human hearts show increased levels of ROS [118]. Aging and oxidative stress can be accompanied by other pathological conditions such as diabetes, endothelial dysfunction, atherosclerosis, hypertension, and degenerative diseases increasing the imbalance between ROS production and antioxidant systems [119]. With aging, the compensatory mechanisms do not effectively manage ROS accumulation in mice models. This generates increased oxidation of proteins, lipids, and mitochondrial DNA damage. Electron leak in mitochondria is considered one of the main sources of ROS and adenine nucleotide translocase (ANT) seems to be why [120,121]. ANT oxidative and carbonyl modifications reduce mitochondrial energy output [122-124]. The uncoupling of ATP synthesis disrupts the mitochondrial matrix disrupting Ca
In the failing aging heart, oxidative stress plays an important role. Both normal aging and pathological aging human hearts show increased levels of ROS[118]. Aging and oxidative stress can be accompanied by other pathological conditions such as diabetes, endothelial dysfunction, atherosclerosis, hypertension, and degenerative diseases increasing the imbalance between ROS production and antioxidant systems[119]. With aging, the compensatory mechanisms do not effectively manage ROS accumulation in mice models. This generates increased oxidation of proteins, lipids, and mitochondrial DNA damage. Electron leak in mitochondria is considered one of the main sources of ROS and adenine nucleotide translocase (ANT) seems to be why[120][121]. ANT oxidative and carbonyl modifications reduce mitochondrial energy output[122][123][124]. The uncoupling of ATP synthesis disrupts the mitochondrial matrix disrupting Ca
2+ stabilization [125,126]. In fact, failing senescent mice hearts in conditions of stress, increase by two-fold Ca
stabilization[125][126]. In fact, failing senescent mice hearts in conditions of stress, increase by two-fold Ca
2+ levels, increasing ischemic and reperfusion damage [127]. Progression to heart failure and heart failure itself includes various mechanisms. As seen before, oxidative stress in aging also leads to increased cardiomyocytes apoptosis, ECM remodeling, and altered response to stress all important factors for HF [128]. For all the reasons above, heart failure and aging are synonyms. The aging process and all the changes involved lead to heart failure.
levels, increasing ischemic and reperfusion damage[127]. Progression to heart failure and heart failure itself includes various mechanisms. As seen before, oxidative stress in aging also leads to increased cardiomyocytes apoptosis, ECM remodeling, and altered response to stress all important factors for HF[128]. For all the reasons above, heart failure and aging are synonyms. The aging process and all the changes involved lead to heart failure.
2.3. Valvular Heart Disease
3.3. Valvular Heart Disease
Age is the main driving factor for valvular myxomatous degeneration and valvular sclerosis. Elderly patients have a prevalence of 30–80% of aortic valve sclerosis[129][130]. With age echocardiographic examination in these patients shows an increase in calcification of the aortic valve leaflets and anulus[131][132]. Elderly patients also have concomitant risk factors for rapid progression of valve degeneration such as hypertension, LVH, hyperlipidemia, and kidney failure[133]. Moreover, there is a linear correlation between aortic valve degeneration and atherosclerosis in elderly patients[134]. Valve sclerosis does not have a hemodynamic impact but can evolve into stenosis. Aortic stenosis is characterized by the reduction in the opening of the valve leaflets with consequent increased in pressure gradient between the left ventricle and the aorta[135]. To guarantee the necessity of the increased pressure gradient the heart undergoes myocardial hypertrophy to maintain an adequate systolic function and cardiac output[136]. This mechanism of compensation leads to left ventricle dilatation and deterioration of systolic function on the long run. Another aortic valve degeneration consequence is aortic regurgitation, a situation where the leaflets do not close properly and a backward blood flow in generated. This second condition causes a rapid diastolic filling and eccentric hypertrophy of the heart in order to increase the blood volume output[137]. Same is true for the mitral valve, with aging there is degenerative process involving the annulus and the leaflets[138][139]. In this case ,however, valve regurgitation is more common than stenosis[140]. As for the aortic valve, hypertension, kidney failure, and aortic alterations are risk factors mitral calcification and patients with this such alteration have an increased risk for heart failure, atrial fibrillation, stroke, coronary artery diseases, and overall adverse cardiovascular events and mortality[141]. In humans, mitral valve regurgitation is mainly associated with ischemic heart disease and myxomatous degeneration while mitral stenosis is associated with rheumatic disease[142]. Mitral and aortic valve vices are one of the most common cause of surgery in older population[143]. Interesting are the recent discoveries that associated reduction in telomere length with degenerative valve disease. In particular elderly patients with aortic stenosis had a reduction in leukocyte telomere length[144] Although more research is required to fully understand ROS-induced damage to telomeric DNA, studies suggests that this may be an important factor to take into consideration[145]. Furthermore, even if oxidative stress and telomere shortening in humans still have not been directly correlated, there is a great association with frailty typical of the elderly[146]. Oxidative stress and inflammation play an important role in valvular calcification. The underlining mechanisms are not fully known but local inflammation by hyperlipidemia and diabetes has been shown to be a great promoter of valve and vascular sclerosis[147] . Moreover, human studies have assessed that oxidized lipids seem to be one of the main inflammatory driving factors[148][149][150]. Increased levels of circulating oxidized phospholipid, ox-LDL and of TNF-alpha and inflammatory cells have been highlighted in patients with higher degrease of valvular degeneration151][152]. High levels of ox-LDL increase the expression of osteogenic factors such as BMP-2, activating TLR expression[153][154]. The vicious cycle of lipid oxidation induces chemokines release and recruitment of monocytes and enhanced expression of ICAM, and MMP, factors leading to reduction NO production[155][156][157][158][159]. In conclusion, valvular heart disease is probably associated to oxidative stress in the aging heart by ROS increase with NO synthase uncoupling mediated mainly by ox-LDL expression of NADPH[160][161].
Age is t The main driving factor for valvular myxomatous degeneration and valvular sclerosis. Elderly patients have a prevalence of 30–80% of aortic valve sclerosis [129,130]. With age echocardiographic exas entry is fromination in these patients shows an increase in calcification of the aortic valve leaflets and anulus [131,132]. Elderly httpatients also have concomitant risk factors for rapid progression of valve degeneration such as hypertension, LVH, hyperlipidemia, and kidney failure [133]. Moreover, there is a linear correlation bet://ween aortic valve degeneration and atherosclerosis in elderly patients [134]. Valve sclerosis does not have a hemodynamic impact but can evolve into stenosis. Aortic stenosis is characterized by the reduction in the opening of the valve leaflets with consequent increased in pressure gradient between the left ventricle and the aorta [135]. To guarantee the necessity of the increased pressure gradient the heart undergoes myocardial hypertrophy to maintain an adequate systolic function and cardiac output [136]dpi. This mechanism of compensation leads to left ventricle dilatation and deterioration of systolic function on the long run. Another aortic valve degeneration consequence is aortic regurgitation, a situation where the leaflets do not close properly and a backward blood flow in generated. This second condition causes a rapid diastolic filling and eccentric hypertrophy of the heart in order to increase the blood volume output [137]. Same is true for the mitral valve, with aging there is degenerative process involving the annulus and the leaflets [138,139]. In this case ,however, valve regurgitation is more common than stenosis [14om/20]. As for the aortic valve, hypertension, kidney failure, and aortic alterations are risk factors mitral calcification and patients with this such alteration have an increased risk for heart failure, atrial fibrillation, stroke, coronary artery diseases, and overall adverse cardiovascular events and mortality [141]. In humans, mitral valve regurgitation is mainly associated with ischemic heart disease and myxomatous degeneration while mitral stenosis is associated with rheumatic disease [142]. Mitral and aortic valve vices are one of the most common cause of surgery in older population [143]. Interesting are the recent discoveries that associated reduction in telomere length with degenerative valve disease. In particular elderly patients with aortic stenosis had a reduction in leukocyte telomere length [144]. Although more research is required to fully understand ROS-induced damage to telomeric DNA, studies suggests that this may be an important factor to take into consideration [145]. Furthermore, even if oxidative stress and telomere shortening in humans still have not been directly correlated, there is a great association with frailty typical of the elderly [146]. Oxidative stress and inflammation play an important role in valvular calcification. The underlining mechanisms are not fully known but local inflammation by hyperlipidemia and diabetes has been shown to be a great promoter of valve and vascular sclerosis [147]. Moreover, human studies have assessed that oxidized lipids seem to be one of the main inflammatory driving factors [148-150]. Increased levels of circulating oxidized phospholipid, ox-LDL and of TNF-alpha and inflammatory cells have been highlighted in patients with higher degrease of valvular degeneration [151,1572]. High levels of ox-LDL increase the expression of osteogenic factors such as BMP-2, activating TLR expression [9/153,154]. The vicious cycle of lipid oxidation induces chemokines release and recruitment of monocytes and enhanced expression of ICAM, and MMP, factors leading to reduction NO production [/155-159]. In conclusion, valvular /60/heart disease is probably associated to oxidative stress in the aging heart by ROS increase with NO synthase uncoupling mediated mainly by ox-LDL expression of NADPH [160,161].
References
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. Mol. Med. 2010, 88, 993–1001.
- Neubauer, S. The failing heart—An engine out of fuel. Engl. J. Med. 2007, 356, 1140–1151.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Harman, D. The biologic clock: The mitochondria? Am. Geriatr. Soc. 1972, 20, 145–147.
- Garinis, G.A.; van der Horst, G.T.; Vijg, J.; Hoeijmakers, J.H. DNA damage and ageing: New-age ideas for an age-old problem. Cell Biol. 2008, 10, 1241–1247.
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Gerontol. 2010, 45, 478–488.
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249.
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Uracil in DNA—General mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516.
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxidative Cell. Longev. 2012, 2012, 282438.
- Dai, D.F.; Rabinovitch, P.S. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009, 19, 213–220.
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Brüning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Genet. 1999, 21, 133–137.
- Wang, J.; Silva, J.P.; Gustafsson, C.M.; Rustin, P.; Larsson, N.G. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Natl. Acad. Sci. USA 2001, 98, 4038–4043.
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Genet. 2008, 40, 392–394.
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell 2009, 10, 131–138.
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metab. 2015, 21, 667–677.
- Kujoth, G.C.; Leeuwenburgh, C.; Prolla, T.A. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006, 66, 7386–7389.
- Tower, J. Programmed cell death in aging. Ageing Res. Rev. 2015, 23, 90–100.
- Modrego, J.; de las Heras, N.; Zamorano-León, J.J.; Mateos-Cáceres, P.J.; Martín-Fernández, B.; Valero-Muñoz, M.; Lahera, V.; López-Farré, A.J. Changes in cardiac energy metabolic pathways in overweighed rats fed a high-fat diet. J. Nutr. 2013, 52, 847–856.
- Opie, L.H.; Knuuti, J. The adrenergic-fatty acid load in heart failure. Am. Coll. Cardiol. 2009, 54, 1637–1646.
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Res. 2012, 93, 79–88.
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 423–430.
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Redox Signal. 2010, 12, 503–535.
- Upadhya, B.; Taffet, G.E.; Cheng, C.P.; Kitzman, D.W. Heart failure with preserved ejection fraction in the elderly: Scope of the problem. Mol. Cell. Cardiol. 2015, 83, 73–87.
- Campbell, S.G.; Haynes, P.; Kelsey Snapp, W.; Nava, K.E.; Campbell, K.S. Altered ventricular torsion and transmural patterns of myocyte relaxation precede heart failure in aging F344 rats. J. Physiol. Heart Circ. Physiol. 2013, 305, 676–686.
- Boluyt, M.O.; Converso, K.; Hwang, H.S.; Mikkor, A.; Russell, M.W. Echocardiographic assessment of age-associated changes in systolic and diastolic function of the female F344 rat heart. Appl. Physiol. 2004, 96, 822–828.
- Maruyama, Y. Aging and arterial-cardiac interactions in the elderly. J. Cardiol. 2012, 155, 14–19.
- Juhaszova, M.; Rabuel, C.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Protection in the aged heart: Preventing the heart-break of old age? Res. 2005, 66, 233–244.
- Mirza, M.; Strunets, A.; Shen, W.K.; Jahangir, A. Mechanisms of arrhythmias and conduction disorders in older adults. Geriatr. Med. 2012, 28, 555–573.
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012, 8, 143–164.
- Dhahbi, J.M.; Tsuchiya, T.; Kim, H.J.; Mote, P.L.; Spindler, S.R. Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 218–231.
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Rev. 1993, 73, 413–467.
- Coleman, R.; Silbermann, M.; Gershon, D.; Reznick, A.Z. Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology 1987, 33, 34–39.
- Muscari, C.; Finelli, C.; Stefanelli, C.; Flamigni, F.; Guarnieri, C.; Caldarera, C.M. Age-dependent differences of ATP breakdown and ATP-catabolite release in ischemic and reperfused hearts. Ageing Dev. 1993, 67, 1–11.
- Muscari, C.; Caldarera, C.M.; Guarnieri, C. Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res. Cardiol. 1990, 85, 172–178.
- Sohal, R.S.; Sohal, B.H. Hydrogen peroxide release by mitochondria increases during aging. Ageing Dev. 1991, 57, 187–202.
- Liu, F.; Li, N.; Long, B.; Fan, Y.Y.; Liu, C.Y.; Zhou, Q.Y.; Murtaza, I.; Wang, K.; Li, P.F. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. 2014, 5, e1171.
- Fajemiroye, J.O.; da Cunha, L.C.; Saavedra-Rodriguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourao, A.A.; da Silva, E.F.; Williams, N.E.E.; Martins, J.L.R.; Sousa, R.B.; et al. Aging-Induced Biological Changes and Cardiovascular Diseases. Res. Int. 2018, 2018, 7156435.
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Res. 1991, 68, 1560–1568.
- Hees, P.S.; Fleg, J.L.; Lakatta, E.G.; Shapiro, E.P. Left ventricular remodeling with age in normal men versus women: Novel insights using three-dimensional magnetic resonance imaging. J. Cardiol. 2002, 90, 1231–1236.
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.R.; Anversa, P. Gender differences and aging: Effects on the human heart. Am. Coll. Cardiol. 1995, 26, 1068–1079.
- Kajstura, J.; Cheng, W.; Sarangarajan, R.; Li, P.; Li, B.; Nitahara, J.A.; Chapnick, S.; Reiss, K.; Olivetti, G.; Anversa, P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. J. Physiol. 1996, 271, 1215–1228.
- Sheydina, A.; Riordon, D.R.; Boheler, K.R. Molecular mechanisms of cardiomyocyte aging. Sci. 2011, 121, 315–329.
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Redox Signal. 2012, 16, 1492–1526.
- Leon, L.J.; Gustafsson, A.B. Staying young at heart: Autophagy and adaptation to cardiac aging. Mol. Cell. Cardiol. 2016, 95, 78–85.
- AlGhatrif, M.; Lakatta, E.G. The conundrum of arterial stiffness, elevated blood pressure, and aging. Hypertens. Rep. 2015, 17, 12.
- Gerstenblith, G.; Frederiksen, J.; Yin, F.C.; Fortuin, N.J.; Lakatta, E.G.; Weisfeldt, M.L. Echocardiographic assessment of a normal adult aging population. Circulation 1977, 56, 273–278.
- Schulman, S.P.; Lakatta, E.G.; Fleg, J.L.; Lakatta, L.; Becker, L.C.; Gerstenblith, G. Age-related decline in left ventricular filling at rest and exercise. J. Physiol. 1992, 263, 1932–1938.
- Miller, T.R.; Grossman, S.J.; Schectman, K.B.; Biello, D.R.; Ludbrook, P.A.; Ehsani, A.A. Left ventricular diastolic filling and its association with age. J. Cardiol. 1986, 58, 531–535.
- Howden, E.J.; Carrick-Ranson, G.; Sarma, S.; Hieda, M.; Fujimoto, N.; Levine, B.D. Effects of Sedentary Aging and Lifelong Exercise on Left Ventricular Systolic Function. Sci. Sports Exerc. 2018, 50, 494–501.
- Peverill, R.E. Aging and the relationships between long-axis systolic and early diastolic excursion, isovolumic relaxation time and left ventricular length-Implications for the interpretation of aging effects on e. PLoS ONE 2019, 14, e0210277.
- Fleg, J.L.; O’Connor, F.; Gerstenblith, G.; Becker, L.C.; Clulow, J.; Schulman, S.P.; Lakatta, E.G. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. Appl. Physiol. 1995, 78, 890–900.
- Silaghi, A.; Piercecchi-Marti, M.D.; Grino, M.; Leonetti, G.; Alessi, M.C.; Clement, K.; Dadoun, F.; Dutour, A. Epicardial adipose tissue extent: Relationship with age, body fat distribution, and coronaropathy. Obesity 2008, 16, 2424–2430.
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. Mol. Cell. Cardiol. 2016, 93, 175–185.
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173.
- Jugdutt, B.I.; Jelani, A.; Palaniyappan, A.; Idikio, H.; Uweira, R.E.; Menon, V.; Jugdutt, C.E. Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model: Effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation 2010, 122, 341–351.
- Mendes, A.B.; Ferro, M.; Rodrigues, B.; Souza, M.R.; Araujo, R.C.; Souza, R.R. Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Medicina 2012, 72, 216–220.
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. Am. Coll. Cardiol. 2008, 51, 802–809.
- Everett, T.H.T.; Li, H.; Mangrum, J.M.; McRury, I.D.; Mitchell, M.A.; Redick, J.A.; Haines, D.E. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation 2000, 102, 1454–1460.
- Elliott, H.L.; Sumner, D.J.; McLean, K.; Reid, J.L. Effect of age on the responsiveness of vascular alpha-adrenoceptors in man. Cardiovasc. Pharmacol. 1982, 4, 388–392.
- Vaitkevicius, P.V.; Fleg, J.L.; Engel, J.H.; O’Connor, F.C.; Wright, J.G.; Lakatta, L.E.; Yin, F.C.; Lakatta, E.G. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 1993, 88, 1456–1462.
- Egashira, K.; Inou, T.; Hirooka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Kuga, T.; Urabe, Y.; Takeshita, A. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation 1993, 88, 77–81.
- Taddei, S.; Virdis, A.; Mattei, P.; Ghiadoni, L.; Gennari, A.; Fasolo, C.B.; Sudano, I.; Salvetti, A. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation 1995, 91, 1981–1987.
- Hotta, H.; Uchida, S. Aging of the autonomic nervous system and possible improvements in autonomic activity using somatic afferent stimulation. Gerontol. Int. 2010, 10, 127–136.
- Niu, S.W.; Huang, J.C.; Chen, S.C.; Lin, H.Y.; Kuo, I.C.; Wu, P.Y.; Chiu, Y.W.; Chang, J.M. Association between Age and Changes in Heart Rate Variability after Hemodialysis in Patients with Diabetes. Aging Neurosci. 2018, 10, 43.
- Bonnemeier, H.; Richardt, G.; Potratz, J.; Wiegand, U.K.; Brandes, A.; Kluge, N.; Katus, H.A. Circadian profile of cardiac autonomic nervous modulation in healthy subjects: Differing effects of aging and gender on heart rate variability. Cardiovasc. Electrophysiol. 2003, 14, 791–799.
- Britton, A.; Shipley, M.; Malik, M.; Hnatkova, K.; Hemingway, H.; Marmot, M. Changes in heart rate and heart rate variability over time in middle-aged men and women in the general population (from the Whitehall II Cohort Study). J. Cardiol. 2007, 100, 524–527.
- Lipsitz, L.A.; Mietus, J.; Moody, G.B.; Goldberger, A.L. Spectral characteristics of heart rate variability before and during postural tilt. Relations to aging and risk of syncope. Circulation 1990, 81, 1803–1810.
- Kanegusuku, H.; Queiroz, A.C.; Silva, V.J.; de Mello, M.T.; Ugrinowitsch, C.; Forjaz, C.L. High-Intensity Progressive Resistance Training Increases Strength With No Change in Cardiovascular Function and Autonomic Neural Regulation in Older Adults. Aging Phys. Act. 2015, 23, 339–345.
- Masuki, S.; Morikawa, M.; Nose, H. Interval Walking Training Can Increase Physical Fitness in Middle-Aged and Older People. Sport Sci. Rev. 2017, 45, 154–162.
- Guedes, J.M.; Pieri, B.; Luciano, T.F.; Marques, S.O.; Guglielmo, L.G.A.; Souza, C.T. Muscular resistance, hypertrophy and strength training equally reduce adiposity, inflammation and insulin resistance in mice with diet-induced obesity. Einstein 2020, 18, eAO4784.
- Masuda, M.; Yamada, T.; Mizuno, H.; Minamiguchi, H.; Konishi, S.; Ohtani, T.; Yamaguchi, O.; Okuyama, Y.; Uematsu, M.; Sakata, Y. Impact of atrial fibrillation ablation on cardiac sympathetic nervous system in patients with and without heart failure. J. Cardiol. 2015, 199, 65–70.
- Tegene, E.; Tadesse, I.; Markos, Y.; Gobena, T. Prevalence and risk factors for atrial fibrillation and its anticoagulant requirement in adults aged >/=40 in Jimma Town, Southwest Ethiopia: A community based cross-sectional study. J. Cardiol. Heart Vasc. 2019, 22, 199–204.
- Xi, Y.; Cheng, J. Dysfunction of the autonomic nervous system in atrial fibrillation. Thorac. Dis. 2015, 7, 193–198.
- Stavrakis, S.; Nakagawa, H.; Po, S.S.; Scherlag, B.J.; Lazzara, R.; Jackman, W.M. The role of the autonomic ganglia in atrial fibrillation. JACC Clin. Electrophysiol. 2015, 1, 1–13.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Kaasik, A.; Kuum, M.; Joubert, F.; Wilding, J.; Ventura-Clapier, R.; Veksler, V. Mitochondria as a source of mechanical signals in cardiomyocytes. Res. 2010, 87, 83–91.
- Iribe, G.; Kaihara, K.; Yamaguchi, Y.; Nakaya, M.; Inoue, R.; Naruse, K. Mechano-sensitivity of mitochondrial function in mouse cardiac myocytes. Biophys. Mol. Biol. 2017, 130, 315–322.
- Lakatta, E.G.; Sollott, S.J. Perspectives on mammalian cardiovascular aging: Humans to molecules. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 132, 699–721.
- Lakatta, E.G. Age-associated cardiovascular changes in health: Impact on cardiovascular disease in older persons. Heart Fail. Rev. 2002, 7, 29–49.
- Lakatta, E.G.; Sollott, S.J.; Pepe, S. The old heart: Operating on the edge. In Novartis Foundation Symposium; Chichester: New York, NY, USA; John Wiley: Hoboken, NJ, USA, 1999; Volume 235, pp. 172–196.
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984.
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Redox Signal. 2006, 8, 691–728.
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383.
- Aronow, W.S. Etiology, pathophysiology, and treatment of atrial fibrillation: Part 1. Rev. 2008, 16, 181–188.
- Schueller, P.O.; Steiner, S.; Hennersdorf, M.G. Atrial fibrillation. Mon. Pharm. 2009, 32, 204–210.
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Rev. 2011, 91, 265–325.
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: Implications for management. Circulation 2011, 124, 2264–2274.
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Heart J. 2020.
- Pan, N.H.; Tsao, H.M.; Chang, N.C.; Chen, Y.J.; Chen, S.A. Aging dilates atrium and pulmonary veins: Implications for the genesis of atrial fibrillation. Chest 2008, 133, 190–196.
- Ravassa, S.; Ballesteros, G.; Diez, J. Aging and atrial fibrillation: A matter of fibrosis. Aging (Albany NY) 2019, 11, 9965–9966.
- Ravassa, S.; Lopez, B.; Querejeta, R.; Echegaray, K.; San Jose, G.; Moreno, M.U.; Beaumont, F.J.; Gonzalez, A.; Diez, J. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. Hypertens. 2017, 35, 853–861.
- Zhan, G.; Wenhua, G.; Jie, H.; Xiang, Q.; Lingzhi, C.; Xue, X.; Xing-Xing, C.; Qianli, Z.; Weijian, H.; Hao, Z. Potential roles of circulating matrix metalloproteinase-28 (MMP-28) in patients with atrial fibrillation. Life Sci. 2018, 204, 15–19.
- Lewkowicz, J.; Knapp, M.; Tankiewicz-Kwedlo, A.; Sawicki, R.; Kaminska, M.; Waszkiewicz, E.; Musial, W.J. MMP-9 in atrial remodeling in patients with atrial fibrillation. In Annales de Cardiologie et D'angeiologie; Elsevier Masson: Paris, France, 2015; Volume 64, pp. 285–291.
- Kamkin, A.; Kiseleva, I.; Isenberg, G.; Wagner, K.D.; Gunther, J.; Theres, H.; Scholz, H. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Biophys. Mol. Biol. 2003, 82, 111–120.
- Chang, S.L.; Chen, Y.C.; Chen, Y.J.; Wangcharoen, W.; Lee, S.H.; Lin, C.I.; Chen, S.A. Mechanoelectrical feedback regulates the arrhythmogenic activity of pulmonary veins. Heart 2007, 93, 82–88.
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299.
- Lee, S.H.; Chen, Y.C.; Chen, Y.J.; Chang, S.L.; Tai, C.T.; Wongcharoen, W.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007, 80, 1806–1815.
- Lai, L.P.; Tsai, C.C.; Su, M.J.; Lin, J.L.; Chen, Y.S.; Tseng, Y.Z.; Huang, S.K. Atrial fibrillation is associated with accumulation of aging-related common type mitochondrial DNA deletion mutation in human atrial tissue. Chest 2003, 123, 539–544.
- Tsuboi, M.; Hisatome, I.; Morisaki, T.; Tanaka, M.; Tomikura, Y.; Takeda, S.; Shimoyama, M.; Ohtahara, A.; Ogino, K.; Igawa, O.; et al. Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. J. Clin. Investig. 2001, 31, 489–496.
- Lin, Y.K.; Chen, Y.A.; Lee, T.I.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Aging Modulates the Substrate and Triggers Remodeling in Atrial Fibrillation. J. 2018, 82, 1237–1244.
- McMurray, J.J.; Pfeffer, M.A. Heart failure. Lancet 2005, 365, 1877–1889.
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, 21–181.
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146.
- Orso, F.; Fabbri, G.; Maggioni, A.P. Epidemiology of Heart Failure. Exp. Pharmacol. 2017, 243, 15–33.
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Rev. Cardiol. 2016, 13, 368–378.
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Heart Fail. Rep. 2017, 14, 385–392.
- Goldstein, S. The changing epidemiology of sudden death in heart failure. Heart Fail. Rep. 2004, 1, 93–97.
- Penjaskovic, D.; Sakac, D.; Dejanovic, J.; Zec, R.; Zec Petkovic, N.; Stojsic Milosavljevic, A. Left ventricular diastolic dysfunction in patients with metabolic syndrome. Pregl. 2012, 65, 18–22.
- Cil, H.; Bulur, S.; Turker, Y.; Kaya, A.; Alemdar, R.; Karabacak, A.; Aslantas, Y.; Ekinozu, I.; Albayrak, S.; Ozhan, H.; et al. Impact of body mass index on left ventricular diastolic dysfunction. Echocardiography 2012, 29, 647–651.
- Katz, S.D. Pathophysiology of Chronic Systolic Heart Failure. A View from the Periphery. Am. Thorac. Soc. 2018, 15, 38–41.
- McMurray, J.J. Clinical practice. Systolic heart failure. Engl. J. Med. 2010, 362, 228–238.
- Chatterjee, K. Pathophysiology of systolic and diastolic heart failure. Clin. 2012, 96, 891–899.
- Henes, J.; Rosenberger, P. Systolic heart failure: Diagnosis and therapy. Opin. Anesthesiol. 2016, 29, 55–60.
- Ennezat, P.V.; Le Jemtel, T.H.; Logeart, D.; Marechaux, S. Heart failure with preserved ejection fraction: A systemic disorder. Med. Interne 2012, 33, 370–380.
- Wachter, R. Diastolic heart failure. Internist 2014, 55, 663–668.
- Del Buono, M.G.; Buckley, L.; Abbate, A. Primary and Secondary Diastolic Dysfunction in Heart Failure with Preserved Ejection Fraction. J. Cardiol. 2018, 122, 1578–1587.
- Lodi, R.; Tonon, C.; Calabrese, V.; Schapira, A.H. Friedreich’s ataxia: From disease mechanisms to therapeutic interventions. Redox Signal. 2006, 8, 438–443.
- Kaneto, H.; Katakami, N.; Kawamori, D.; Miyatsuka, T.; Sakamoto, K.; Matsuoka, T.A.; Matsuhisa, M.; Yamasaki, Y. Involvement of oxidative stress in the pathogenesis of diabetes. Redox Signal. 2007, 9, 355–366.
- Bianchi, M.; Panerai, A.E. Formalin injection in the tail facilitates hindpaw withdrawal reflexes induced by thermal stimulation in the rat: Effect of paracetamol. Lett. 1997, 237, 89–92.
- Murphy, M.P. How mitochondria produce reactive oxygen species. J. 2009, 417, 1–13.
- Sohal, R.S.; Arnold, L.A.; Sohal, B.H. Age-related changes in antioxidant enzymes and prooxidant generation in tissues of the rat with special reference to parameters in two insect species. Free Radic. Biol. Med. 1990, 9, 495–500.
- Yan, L.J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Natl. Acad. Sci. USA 1998, 95, 12896–1290.
- Kim, J.H.; Woldgiorgis, G.; Elson, C.E.; Shrago, E. Age-related changes in respiration coupled to phosphorylation. I. Hepatic mitochondria. Ageing Dev. 1988, 46, 263–277.
- Yokozawa, T.; Satoh, A.; Cho, E.J. Ginsenoside-Rd attenuates oxidative damage related to aging in senescence-accelerated mice. Pharm. Pharmacol. 2004, 56, 107–113.
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: Implications for the mitochondrial theory of aging. FASEB J. 2005, 19, 419–421.
- Mo, J.Q.; Hom, D.G.; Andersen, J.K. Decreases in protective enzymes correlates with increased oxidative damage in the aging mouse brain. Ageing Dev. 1995, 81, 73–82.
- Hoffmann, B.; Stockl, A.; Schlame, M.; Beyer, K.; Klingenberg, M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. Biol. Chem. 1994, 269, 1940–1944.
- Nehal, M.; Azam, M.; Baquer, N.Z. Changes in the levels of catecholamines, hexokinase and glucose 6-phosphate dehydrogenase in red cell aging. Int. 1990, 22, 517–522.
- Karavidas, A.; Lazaros, G.; Tsiachris, D.; Pyrgakis, V. Aging and the cardiovascular system. J. Cardiol. 2010, 51, 421–427.
- Nassimiha, D.; Aronow, W.S.; Ahn, C.; Goldman, M.E. Association of coronary risk factors with progression of valvular aortic stenosis in older persons. J. Cardiol. 2001, 87, 1313–1314.
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. Am. Coll. Cardiol. 1997, 29, 630–634.
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation 2005, 111, 3316–3326.
- Otto, C.M.; Lind, B.K.; Kitzman, D.W.; Gersh, B.J.; Siscovick, D.S. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. Engl. J. Med. 1999, 341, 142–147.
- Olsen, M.H.; Wachtell, K.; Bella, J.N.; Gerdts, E.; Palmieri, V.; Nieminen, M.S.; Smith, G.; Ibsen, H.; Devereux, R.B.; Substudy, L. Aortic valve sclerosis relates to cardiovascular events in patients with hypertension (a LIFE substudy). J. Cardiol. 2005, 95, 132–136.
- Otto, C.M. Why is aortic sclerosis associated with adverse clinical outcomes? Am. Coll. Cardiol. 2004, 43, 176–178.
- Faggiano, P.; Antonini-Canterin, F.; Erlicher, A.; Romeo, C.; Cervesato, E.; Pavan, D.; Piazza, R.; Huang, G.; Nicolosi, G.L. Progression of aortic valve sclerosis to aortic stenosis. J. Cardiol. 2003, 91, 99–101.
- Jeon, D.S.; Atar, S.; Brasch, A.V.; Luo, H.; Mirocha, J.; Naqvi, T.Z.; Kraus, R.; Berman, D.S.; Siegel, R.J. Association of mitral annulus calcification, aortic valve sclerosis and aortic root calcification with abnormal myocardial perfusion single photon emission tomography in subjects age < or =65 years old. Am. Coll. Cardiol. 2001, 38, 1988–1993.
- Fulkerson, P.K.; Beaver, B.M.; Auseon, J.C.; Graber, H.L. Calcification of the mitral annulus: Etiology, clinical associations, complications and therapy. J. Med. 1979, 66, 967–977.
- Correia, L.C.; Lakatta, E.G.; O’Connor, F.C.; Becker, L.C.; Clulow, J.; Townsend, S.; Gerstenblith, G.; Fleg, J.L. Attenuated cardiovascular reserve during prolonged submaximal cycle exercise in healthy older subjects. Am. Coll. Cardiol. 2002, 40, 1290–1297.
- Akins, C.W.; Daggett, W.M.; Vlahakes, G.J.; Hilgenberg, A.D.; Torchiana, D.F.; Madsen, J.C.; Buckley, M.J. Cardiac operations in patients 80 years old and older. Thorac. Surg. 1997, 64, 606–614.
- Kurz, D.J.; Kloeckener-Gruissem, B.; Akhmedov, A.; Eberli, F.R.; Buhler, I.; Berger, W.; Bertel, O.; Luscher, T.F. Degenerative aortic valve stenosis, but not coronary disease, is associated with shorter telomere length in the elderly. Thromb. Vasc. Biol. 2006, 26, 114–117.
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Ageing Dev. 2019, 177, 37–45.
- Martinez-Ezquerro, J.D.; Rodriguez-Castaneda, A.; Ortiz-Ramirez, M.; Sanchez-Garcia, S.; Rosas-Vargas, H.; Sanchez-Arenas, R.; Garcia-de la Torre, P. Oxidative Stress, Telomere Length, and Frailty in an Old Age Population. bioRxiv 2019, 71, 414680.
- Cho, K.I.; Sakuma, I.; Sohn, I.S.; Jo, S.H.; Koh, K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018, 277, 60–65.
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Thromb. Vasc. Biol. 2014, 34, 715–723.
- van Dijk, R.A.; Kolodgie, F.; Ravandi, A.; Leibundgut, G.; Hu, P.P.; Prasad, A.; Mahmud, E.; Dennis, E.; Curtiss, L.K.; Witztum, J.L.; et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. Lipid Res. 2012, 53, 2773–2790.
- Ravandi, A.; Leibundgut, G.; Hung, M.Y.; Patel, M.; Hutchins, P.M.; Murphy, R.C.; Prasad, A.; Mahmud, E.; Miller, Y.I.; Dennis, E.A.; et al. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. Am. Coll. Cardiol. 2014, 63, 1961–1971.
- Cote, C.; Pibarot, P.; Despres, J.P.; Mohty, D.; Cartier, A.; Arsenault, B.J.; Couture, C.; Mathieu, P. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 2008, 94, 1175–1180.
- Mohty, D.; Pibarot, P.; Despres, J.P.; Cote, C.; Arsenault, B.; Cartier, A.; Cosnay, P.; Couture, C.; Mathieu, P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Thromb. Vasc. Biol. 2008, 28, 187–193.
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. J. Physiol. Cell Physiol. 2008, 294, 29–35.
- Yang, X.; Fullerton, D.A.; Su, X.; Ao, L.; Cleveland, J.C., Jr.; Meng, X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. Am. Coll. Cardiol. 2009, 53, 491–500.
- Yeang, C.; Wilkinson, M.J.; Tsimikas, S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Opin. Cardiol. 2016, 31, 440–450.
- Murugesan, G.; Sandhya Rani, M.R.; Gerber, C.E.; Mukhopadhyay, C.; Ransohoff, R.M.; Chisolm, G.M.; Kottke-Marchant, K. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. Mol. Cell. Cardiol. 2003, 35, 1375–1384.
- Chang, M.C.; Lee, J.J.; Chen, Y.J.; Lin, S.I.; Lin, L.D.; Jein-Wen Liou, E.; Huang, W.L.; Chan, C.P.; Huang, C.C.; Jeng, J.H. Lysophosphatidylcholine induces cytotoxicity/apoptosis and IL-8 production of human endothelial cells: Related mechanisms. Oncotarget 2017, 8, 106177–106189.
- Rolin, J.; Vego, H.; Maghazachi, A.A. Oxidized lipids and lysophosphatidylcholine induce the chemotaxis, up-regulate the expression of CCR9 and CXCR4 and abrogate the release of IL-6 in human monocytes. Toxins 2014, 6, 2840–2856.
- Inoue, N.; Takeshita, S.; Gao, D.; Ishida, T.; Kawashima, S.; Akita, H.; Tawa, R.; Sakurai, H.; Yokoyama, M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis 2001, 155, 45–52.
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. Am. Coll. Cardiol. 2008, 52, 843–850.
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. Biol. Chem. 2003, 278, 22546–22554.
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Opin. Pharmacol. 2013, 13, 161–167.
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biophys. Res. Commun. 1999, 263, 681–684.
- Rueckschloss, U.; Galle, J.; Holtz, J.; Zerkowski, H.R.; Morawietz, H. Induction of NAD(P)H oxidase by oxidized low-density lipoprotein in human endothelial cells: Antioxidative potential of hydroxymethylglutaryl coenzyme A reductase inhibitor therapy. Circulation 2001, 104, 1767–1772.