Metabolic reprogramming is now recognized as a hallmark of cancer cells and supports cancer growth. Elucidating the underlying mechanisms of metabolic reprogramming in cancer cells may help identifying cancer targets and treatment strategies.
- metabolic reprograming
- cancer metabolism
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Cancer cells need to acquire a different metabolic state than that of non-tumor cells in order to proliferate, invade, and metastasize. During cancer progression, cancer cells encounter various kinds of metabolic stress. First, tumor microenvironments are generally hypoxic and acidic and have a distinct nutrient composition compared to non-tumor tissues from the primary site, which forces cancer cells to adapt in order to grow and invade in these environments. Second, to enter and survive in vessels, cancer cells must reprogram their metabolic state, allowing for anchorage-independent growth that induces extensive oxidative stress in cancer cells. Finally, once cancer cells colonize other organs, they must adapt to quite distinct metabolic environments than those present in primary sites [1]. Overall, because cancer cells need to reprogram their metabolic state during each step of cancer progression, metabolic reprogramming has been recognized as one of the hallmarks of cancer [2].
Elucidating the mechanisms underlying metabolic reprogramming during cancer progression can reveal the metabolic vulnerabilities of cancer cells. This may ultimately result in the identification of new therapeutic targets for cancer and improvement of patients’ prognosis.
2. Metabolic Reprograming during Anchorage-Independent Growth
To form metastases, cancer cells must detach from the ECM and enter and survive in the blood or lymphatic stream, where anchorage-independent growth is required. A very small proportion of circulating cancer cells are capable of surviving and forming metastases [105–107][3][4][5], partly because the metabolic adaptation is required in the process. On top of this, detachment from the ECM induces oxidative stress [108][6], and cancer cells must thus alter their metabolism to respond to this challenge in order to achieve anchorage-independent growth. To maintain redox homeostasis during anchorage-independent growth, tumor spheroids suppress oxidation of glucose and glutamine nutrients, while enhancing reductive formation of citrate from glutamine [109][7]. This reductive carboxylation is dependent on cytosolic IDH1 and results in the suppression of mitochondrial reactive oxygen species (ROS), maximizing anchorage-independent growth (Figure 1) [109][7]. Moreover, fatty acid synthase (FASN) is essential to maintain IDH1-dependent reductive carboxylation of glutamine that quenches excessive ROS produced during the transition from 2D growth to 3D anchorage-independent growth [110][8]. Accordingly, the genetic deletion or pharmacologic inhibition of FASN prevents tumor development and invasive growth, indicating that FASN is a potential target for cancer therapy [110][8]. Another factor that can enhance the survival of cancer cells after detachment from ECM is the ability of these cells to form clusters or aggregates [111,112][9][10]. Cell clustering induces hypoxia, which in turn activates mitophagy to remove damaged mitochondria and reductive carboxylation that supports glycolysis [113][11]. These responses reduce mitochondrial ROS production and promote metastasis [113][11].
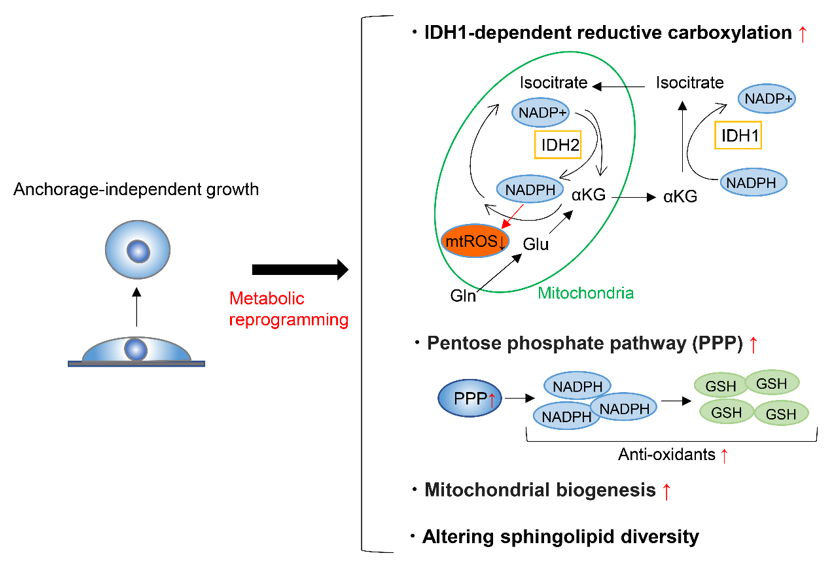
Figure 1. Metabolic reprograming during anchorage-independent growth. Metabolic reprograming during anchorage-independent growth include isocitrate dehydrogenases-1 (IDH1)-dependent reductive carboxylation of glutamine, upregulation of pentose phosphate pathway, the enhancement of mitochondrial biogenesis and respiration, and altering sphingolipid diversity. mtROS; mitochondrial reactive oxygen species, GSH; glutathione-SH.
On the other hand, during tumorigenesis of KRAS-mutant cells, KRAS-induced anchorage-independent growth requires mitochondrial metabolism and ROS production via regulation of the extracellular regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) signaling pathway [114][12]. The major source of ROS generation required for this type of cell growth is the Qo site of mitochondrial complex III. The major function of glucose metabolism during KRAS-induced anchorage-independent growth is to support the pentose phosphate pathway [114][12]. Through gene expression analysis of sorted green fluorescent protein (GFP)-labelled breast cancer cells, circulating cancer cells were shown to exhibit enhanced mitochondria biogenesis and respiration due to upregulation of peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1α) (Figure 1) [115][13]. Moreover, clinical analysis of human breast cancers demonstrates a significant correlation between the PGC-1α expression in invasive cancer cells and the formation of distant metastases [115][13]. In addition, breast cancer cells undergoing anchorage-independent growth exhibit increased mitochondrial biogenesis and upregulation of genes involved in the TCA cycle and pentose phosphate pathway (Figure 1) [116][14]. Overall, these gene signatures predict the potential for metastasis of breast and lung cancer cells [116][14].
Regarding the amino acid metabolism required for anchorage-independent tumor growth, apart from anaplerosis and reductive carboxylation of glutamine, tumor spheroids show increased alanine secretion compared to adherent cells [117][15]. Decreasing alanine secretion by genetical or pharmacological inhibition of mitochondrial pyruvate carrier enhanced spheroid growth and increased serine synthesis, while alanine supplementation had the opposite effect. It was thus suggested that a balance between alanine and serine may regulate spheroid viability through altered sphingolipid diversity (Figure 1) [117][15].
In summary, cancer cells face various stresses during anchorage-independent growth and must reprogram their metabolic state to overcome these stresses. Although the mechanism behind the metabolic reprograming depends on the cancer cell type, most examples include the reductive carboxylation of glutamine that quenches excessive mitochondrial ROS, the upregulation of pentose phosphate pathway, the enhancement of mitochondrial biogenesis and respiration, or the rewiring of a metabolic network involving serine, alanine, and pyruvate that alters sphingolipid diversity. Elucidating specific metabolic reprograming involved in anchorage-independent growth can lead to the identification of therapeutic strategies to prevent cancer metastasis.
3. Metabolic Reprogramming to Form Metastatic Tumors
Metastasis is a major cause of death in cancer patients. Despite various attempts to establish therapeutic strategies, few treatment strategies exist that completely cure patients with metastasis. One reason for the failure of these treatments or prevention strategies is the complex cascade involved in metastatic formation. As mentioned above, cancer cells proliferate and invade the primary tumor site and only then enter the circulation. Finally, cancer cells colonize the metastatic niche and gradually transition to an established metastasis. Cancer cells must then adapt their metabolic state to survive in the metastatic niche where the metabolic environment is distinct from that of the primary site. Elucidating the mechanisms underlying cancer cell adaptation to metastatic niches could lead to the development of new strategies against cancer metastasis. In this section, we summarize the current knowledge on the metabolic pathways and enzymes involved in enabling cancer metastasis.
3.1. Amino Acids
Amino acids are important for the synthesis of proteins, nucleotides, lipids, antioxidants, and TCA cycle intermediates, all of which are needed for cell proliferation and tumor metastasis. For example, endogenously produced or exogenously uptaken asparagine increases the invasive and metastatic potential of breast cancer cells and was shown to upregulate EMT genes (Figure 2A) [118][16]. Moreover, in primary tumors, the expression of asparagine synthetase (ASNS), which generates asparagine from aspartate, correlates with metastatic formation. Indeed, ASNS knockdown and dietary reduction of asparagine suppress breast cancer progression, indicating that ASNS can be a therapeutic target to inhibit metastasis formation in patients. Moreover, high ASNS expression is correlated with poor prognosis in many cancer types [118][16]. Proline catabolism via proline dehydrogenase (PRODH) also enhances growth of breast cancer cells in 3D culture conditions [119][17]. Moreover, PRODH expression and proline catabolism are increased in metastatic tumors compared to primary breast cancers of patients and mice. In fact, PRODH knockdown impairs the capacity of breast cancer cells to form spheroids in vitro and lung metastases in vivo. In addition, the use of the PRODH inhibitor L-THFA is sufficient to impair formation of lung metastases in orthotopic mouse models, indicating that PRODH is a potential drug target for metastasis inhibition in breast cancer patients (Figure 2A) [119][17]. Tumors arising from serine-limited microenvironments in mouse models of melanoma and breast cancer are reported to benefit from upregulated expression of phosphoglycerate dehydrogenase (PHGDH), a component of the serine synthesis pathway that catalyzes the rate-limiting step of glucose-derived serine synthesis [120][18]. The microenvironment of the brain is known to contain lower amounts of serine and glycine than the plasma [121,122][19][20]. Recently, PHGDH was shown to be a major determinant of brain metastasis in multiple human cancer types and preclinical models of breast cancer and melanoma (Figure 2A) [123][21]. In this particular study, genetic and pharmacological inhibition of PHGDH attenuated brain metastasis, indicating that PHGDH inhibitors may be useful in the treatment of brain metastasis [123][21].
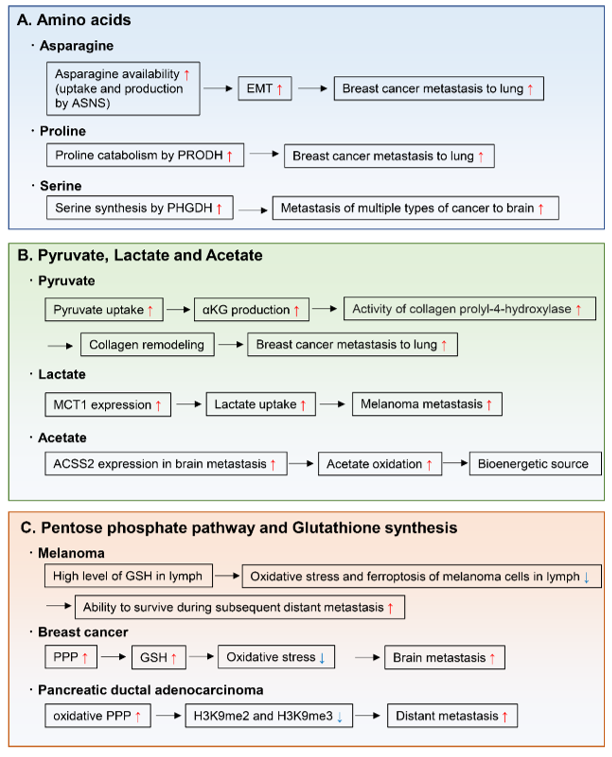
Figure 2. Metabolic reprogramming to form metastatic tumors. The metabolic pathways and enzymes involved in enabling cancer metastasis include those of (A) amino acids, (B) pyruvate, lactate and acetate, and (C) pentose phosphate pathway and glutathione synthesis. ASNS; asparagine synthetase, EMT; epithelial-to-mesenchymal transition, PRODH; proline dehydrogenase, PHGDH; phosphoglycerate dehydrogenase, αKG; α-ketoglutarate, MCT1; monocarboxylate transporter 1, ACSS2; acetyl-CoA synthetase 2, PPP; pentose phosphate pathway, GSH; glutathione-SH.
3.2. Pyruvate, Lactate, and Acetate
Other important nutrients that support tumor metastasis are pyruvate, lactate, and acetate (Figure 2B). During cancer growth in the metastatic niche, cancer cells hydroxylate collagen to form the ECM in the metastatic niche [124][22]. A recent study demonstrated that pyruvate uptake induces the production of αKG and then activates collagen hydroxylation by increasing the activity of the collagen prolyl-4-hydroxylase enzyme [125][23]. Using two different mouse models, this study also showed that inhibition of pyruvate metabolism by pharmacological or genetic inhibition of the pyruvate transporter monocarboxylate transporter 2 is sufficient to decrease collagen hydroxylation and consequently the lung metastases of breast cancer cells [125][23]. Another study showed that after lung metastasis, breast cancer cells convert their metabolic state to pyruvate carboxylase-dependent refilling of the TCA cycle (anaplerosis) because of the enriched availability of pyruvate in the lung environment [126][24].
Lactate has been considered as a waste metabolite that is produced and exported by cells that rely on an active glycolytic system. However, some cancer cells uptake and metabolize lactate in culture [127,128][25][26]. Indeed, studies using stable isotope-labeled lactate show that mouse lung and pancreatic cancers [129][27] and human lung cancers [130][28] use MCT1 to transport lactate from the circulation into the tumor, using part of the carbon from lactate to support the TCA cycle. Of note, during the metastatic process, circulating lactate facilitates the metastatic potential of melanoma cells in patient-derived xenografts and in mouse melanomas [131][29]. Efficient metastatic cells have high-expression levels of MCT1, and inhibition of MCT1 reduces their lactate uptake and metastatic potential [131][29].
Regarding carbon sources for bioenergetics in brain metastases, metastatic mouse orthotopic brain tumors were shown to oxidize acetate in the TCA cycle [132][30]. Moreover, oxidation of acetate was confirmed in patients with brain metastases and was correlated with expression of acetyl-CoA synthetase enzyme 2, which converts acetate to acetyl-CoA [132][30].
3.3. Pentose Phosphate Pathway and Glutathione Synthesis
Increased activity of the pentose phosphate pathway and in glutathione synthesis support the capacity of pancreatic cancer cells to metastasize and proliferate in the liver and lung [133][31], and of breast cancer cells to survive in the brain microenvironment (Figure 2C) [134][32]. This is achieved by the effect of these two pathways in reducing oxidative stress during proliferation in the metastatic organ. As another example, during the evolution of distant metastases, PDAC co-evolved a dependence on the oxidative branch of the pentose phosphate pathway through global epigenetic reprogramming of H3K9 methylation (Figure 2C) [133][31]. The mechanism by which melanoma cells tend to metastasize, initially to the lymph nodes and then to other distant organs, has recently been linked to decreased oxidative stress in the lymphatic environment (Figure 2C) [135][33]. This may be caused by higher levels of glutathione and oleic acid and lower free-iron levels in the lymph, which decrease oxidative stress and ferroptosis. This ultimately contributes to protect melanoma cells from ferroptosis and increases their ability to form metastases in distant organs [135][33].
3.4. Lipid
Lipid metabolism plays an important role in tumor invasion and metastasis [136] [137][34][35]. Cancer cells optimize their requirements for aggressive progression by switching lipid anabolism and catabolism. As for lipid anabolism, fatty acids are indispensable for the biosynthesis of most lipids, and de novo synthesis of fatty acids has been shown to drive tumor progression [138,139][36][37]. Fatty acids synthesis relies on the activation of the fatty acid biosynthetic enzymes, adenosine triphosphate citrate lyase (ACLY), acetyl-CoA carboxylase (ACC) and FASN. ACLY is responsible for de novo fatty acid synthesis as a key enzyme by converting citrate to oxaloacetate and cytosolic acetyl-CoA. Recent study showed that ACLY promotes colon cancer cell metastasis by stabilizing β-catenin 1 protein [140][38]. ACC is the rate-limiting enzyme in the fatty acid synthesis pathway, which carboxylates acetyl-CoA to form malonyl-CoA. High phospho-ACC expression was shown to be independently associated with worse overall survival in patients with lymph nodes metastasis of squamous cell carcinoma of the head and neck [141][39]. In hepatocellular carcinoma (HCC) patients, up-regulation of ACC1 was associated with multiple aggressive characteristics of HCC, such as vascular invasion and poor differentiation [142][40]. FASN produces saturated fatty acids, which uses one acetyl-CoA and sequentially adds seven malonyl-CoA molecules to form the 16-carbon palmitate. As an example of FASN promoting cancer metastasis, FASN expression enhanced peritoneal metastasis of ovarian cancer in part through the induction of epithelial mesenchymal transition (EMT) [143][41].
Fatty acids are catabolized by the fatty acid oxidation (FAO), also known as β oxidation, which sustain ATP levels and NADPH production. Thus, FAO contributes to energy production and redox homeostasis. Recent studies suggest that FAO is an important energy pathway in metastatic triple-negative breast cancer (TNBC) [144,145][42][43]. CUB-domain containing protein 1 (CDCP1) regulates lipid metabolism by decreasing cytoplasmic lipid droplet abundance, stimulating fatty acid oxidation and oxidative phosphorylation [144][42]. This metabolic reprogramming contributes to the energy production required for cell migration and metastasis of TNBC. The other study showed that metastatic TNBC maintained high levels of ATP through FAO and activated Src oncoprotein, which promoted TNBC metastasis [145][43].
Uptake of exogenous fatty acids into cancer cells could facilitate metastasis [138][36]. Fatty acids are translocated across the plasma membrane through either passive diffusion or a transport system. CD36 is one of the most well-characterized receptors of fatty acids in cancer cells. Recent study showed that CD36 expression in human oral carcinoma cells was essential for metastasis [146][44]. Mechanically, CD36-expressing metastatic cells utilize fatty acid oxidation and obtain energy required for them to survive at metastatic sites. Another study showed that adipocytes induced CD36 expression in ovarian cancer cells, which promoted metastasis of cancer cells in peritoneum and omentum [147][45]. And also, CD36 was shown to be a key mediator of fatty acids-induced metastasis of gastric cancer cells via the AKT/glycogen synthase kinase (GSK)-3β/β-catenin signaling pathway [148][46].
References
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354.
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308.
References
- Schild, T.; Low, V.; Blenis, J.; Gomes, A.P. Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell 2018, 33, 347–354.
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308.
- Stott, S.L.; Richard, L.; Nagrath, S.; Min, Y.; Miyamoto, D.T.; Ulkus, L.; Inserra, E.J.; Ulman, M.; Springer, S.; Nakamura, Z.; et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2010, 2, 25ra23.
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239.
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258.
- Bueno, M.J.; Jimenez-Renard, V.; Samino, S.; Capellades, J.; Junza, A.; López-Rodríguez, M.L.; Garcia-Carceles, J.; Lopez-Fabuel, I.; Bolaños, J.P.; Chandel, N.S.; et al. Essentiality of fatty acid synthase in the 2D to anchorage-independent growth transition in transforming cells. Nat. Commun. 2019, 10, 5011.
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122.
- Labuschagne, C.F.; Cheung, E.C.; Blagih, J.; Domart, M.C.; Vousden, K.H. Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab. 2019, 30, 720–734.
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793.
- Lebleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; De Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003.
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805.
- Muthusamy, T.; Cordes, T.; Handzlik, M.K.; You, L.; Lim, E.W.; Gengatharan, J.; Pinto, A.F.M.; Badur, M.G.; Kolar, M.J.; Wallace, M.; et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature 2020, 586, 790–795.
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381.
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267.
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4.
- Rainesalo, S.; Keränen, T.; Palmio, J.; Peltola, J.; Oja, S.S.; Saransaari, P. Plasma and Cerebrospinal Fluid Amino Acids in Epileptic Patients. Neurochem Res. 2004, 29, 319–324.
- Dolgodilina, E.; Imobersteg, S.; Laczko, E.; Welt, T.; Verrey, F.; Makrides, V. Brain interstitial fluid glutamine homeostasis is controlled by amino acid transporter. J. Cereb. Blood Flow Metab. 2016, 36, 1929–1941.
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373.
- Gilkes, D.M.; Chaturvedi, P.; Bajpai, S.; Wong, C.C.; Wei, H.; Pitcairn, S.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Collagen Prolyl Hydroxylases Are Essential for Breast Cancer Metastasis. Cancer Res. 2013, 73, 3285–3297.
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121.
- Christen, S.; Lorendeau, D.; Schmieder, R.; Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848.
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66.
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942.
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118.
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120.
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614.
- Mcdonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376.
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Iii, J.R.Y.; Felding-habermann, B. Adaptation of Energy Metabolism in Breast Cancer Brain Metastases. Cancer Res. 2007, 67, 1472–1486.
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118.
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76.
- Peck, B.; Schulze, A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer 2019, 5, 693–703.
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22.
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777.
- Wen, J.; Min, X.; Shen, M.; Hua, Q.; Han, Y.; Zhao, L.; Liu, L.; Huang, G.; Liu, J.; Zhao, X. ACLY facilitates colon cancer cell metastasis by CTNNB1. J. Exp. Clin. Cancer Res. 2019, 38, 401.
- Su, Y.W.; Lin, Y.H.; Pai, M.H.; Lo, A.C.; Lee, Y.C.; Fang, I.C.; Lin, J.; Hsieh, R.K.; Chang, Y.F.; Chen, C.L. Association between phosphorylated AMP-activated protein kinase and acetyl-CoA carboxylase expression and outcome in patients with squamous cell carcinoma of the head and neck. PLoS ONE 2014, 9.
- Wang, M.D.; Wu, H.; Fu, G.B.; Zhang, H.L.; Zhou, X.; Tang, L.; Dong, L.W.; Qin, C.J.; Huang, S.; Zhao, L.H.; et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 2016, 63, 1272–1286.
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int. J. Mol. Sci. 2014, 15, 11539–11554.
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565.
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165.
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45.
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301.
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–15.