Drug therapy in pediatric patients is challenging in view of the maturation of organ systems and processes that affect pharmacokinetics and pharmacodynamics. Especially for the youngest age groups and for pediatric-only indications, neonatal and juvenile animal models can be useful to assess drug safety and to better understand the mechanisms of diseases or conditions. In this respect, the use of neonatal and juvenile pigs in the field of pediatric drug discovery and development is promising, although still limited at this point. This review summarizes the comparative postnatal development of pigs and humans and discusses the advantages of the juvenile pig in view of developmental pharmacology, pediatric diseases, drug discovery and drug safety testing. Furthermore, limitations and unexplored aspects of this large animal model are covered. At this point in time, the potential of the neonatal and juvenile pig as nonclinical safety models for pediatric drug development is underexplored.
Drug therapy in pediatric patients is challenging in view of the maturation of organ systems and processes that affect pharmacokinetics and pharmacodynamics. Especially for the youngest age groups and for pediatric-only indications, neonatal and juvenile animal models can be useful to assess drug safety and to better understand the mechanisms of diseases or conditions. In this respect, the use of neonatal and juvenile pigs in the field of pediatric drug discovery and development is promising, although still limited at this point. This study summarizes the comparative postnatal development of pigs and humans and discusses the advantages of the juvenile pig in view of developmental pharmacology, pediatric diseases, drug discovery and drug safety testing. Furthermore, limitations and unexplored aspects of this large animal model are covered. At this point in time, the potential of the neonatal and juvenile pig as nonclinical safety models for pediatric drug development is underexplored.
- pediatric pharmacology
- juvenile pig model
- translational research
- drug discovery
- drug development
- drug safety
- PBPK
1. Introduction
Clinical pharmacology aims to evaluate and understand drug-specific (side)-effects based on pharmacokinetics (PK) and pharmacodynamics (PD). Pharmacokinetics (absorption, distribution, metabolism and excretion; ADME) describes drug concentration time courses in a given compartment, like blood, cerebrospinal fluid or subcutaneous tissues. When considering the systemic circulation, the concentration time course is dictated by volume of distribution (Vd) and clearance (CL) as primary PK parameters. Pharmacodynamics describes the link between drug concentrations (in the systemic circulation and/or at the site of action) and effects or side-effects over time. Throughout pediatric life, it is reasonable to anticipate that both PK as well as PD are affected by developmental changes, reflecting both maturation and growth or weight/size changes [1].
All ADME processes display maturational changes, so that extrapolation and dosing based on simple linear (e.g.,/kg or/BSA) or allometric scaling (kg
x) commonly results in over- or under-exposure in specific subpopulations [2]. Besides the physicochemical properties of a given drug, the rate (and extent) of absorption (typically represented by an absorption rate constant) is affected by developmental physiology, i.e., gastric emptying time, pH or gastric fluid volume; intestinal transit time, bile concentrations or volumes; first pass effect factors like drug-metabolizing enzymes or drug transporters [3]. As an illustration, the oral bioavailability of midazolam is highest in preterm neonates (49–92%), 25–85% in infants and 21–30% after infancy, mirroring ontogeny of the activity of enzymes involved in first pass metabolism [4]. The same holds true for maturational patterns on distribution. After reaching the systemic circulation, a drug will distribute to different compartments (tissues, organs). This distribution behavior is determined by the physicochemical properties of the drug (size, degree of ionization at physiological pH, lipophilicity or water solubility), but further depends on maturational changes such as cardiac output and organ-specific blood flow, and body and plasma composition, as well as the interaction between them, e.g., plasma and tissue protein binding kinetics. To illustrate this, the body water/kg body weight proportion is up to 80–90% in preterm neonates, with a subsequent decrease to 60% after infancy [5]. Protein binding capacity is dependent on maturational plasma protein binding capacity, as illustrated for, e.g., vancomycin [6]. The liver, followed by the small intestine and the kidneys, are the major sites of drug metabolism. In the liver, changes in activity for both phase I and II enzymes are observed in an iso-enzyme-specific manner [1,7]. Maturational changes in these organs will be further discussed later in this review. Excretion is mainly by the kidney via glomerular filtration and tubular secretion and reabsorption, but occasionally also occurs by hepato-biliary or respiratory routes. Similar to hepatic metabolism, each of these processes displays characteristic scenarios of maturation. Compared to glomerular filtration, tubular functions mature more slowly, with tubular secretion at adult equivalent level by 15 months, and tubular reabsorption by 24 months [8,9]. Maturational aminoglycoside clearance closely follows the development of glomerular filtration, while digoxin clearance is, in part, affected by the maturation of tubular secretion capacity [10,11]. These PK profiles are further affected by a diverse set of nonmaturational covariates, including but not limited to genetics, disease characteristics (e.g., obesity, chronic inflammation, critical illness, chronic kidney disease, asphyxia) or environmental factors (e.g., type of nutrition, comedication, drug formulation, treatment modalities like therapeutic hypothermia) [12].
) commonly results in over- or under-exposure in specific subpopulations [2]. Besides the physicochemical properties of a given drug, the rate (and extent) of absorption (typically represented by an absorption rate constant) is affected by developmental physiology, i.e., gastric emptying time, pH or gastric fluid volume; intestinal transit time, bile concentrations or volumes; first pass effect factors like drug-metabolizing enzymes or drug transporters [3]. As an illustration, the oral bioavailability of midazolam is highest in preterm neonates (49–92%), 25–85% in infants and 21–30% after infancy, mirroring ontogeny of the activity of enzymes involved in first pass metabolism [4]. The same holds true for maturational patterns on distribution. After reaching the systemic circulation, a drug will distribute to different compartments (tissues, organs). This distribution behavior is determined by the physicochemical properties of the drug (size, degree of ionization at physiological pH, lipophilicity or water solubility), but further depends on maturational changes such as cardiac output and organ-specific blood flow, and body and plasma composition, as well as the interaction between them, e.g., plasma and tissue protein binding kinetics. To illustrate this, the body water/kg body weight proportion is up to 80–90% in preterm neonates, with a subsequent decrease to 60% after infancy [5]. Protein binding capacity is dependent on maturational plasma protein binding capacity, as illustrated for, e.g., vancomycin [6]. The liver, followed by the small intestine and the kidneys, are the major sites of drug metabolism. In the liver, changes in activity for both phase I and II enzymes are observed in an iso-enzyme-specific manner [1][7]. Maturational changes in these organs will be further discussed later in this review. Excretion is mainly by the kidney via glomerular filtration and tubular secretion and reabsorption, but occasionally also occurs by hepato-biliary or respiratory routes. Similar to hepatic metabolism, each of these processes displays characteristic scenarios of maturation. Compared to glomerular filtration, tubular functions mature more slowly, with tubular secretion at adult equivalent level by 15 months, and tubular reabsorption by 24 months [8][9]. Maturational aminoglycoside clearance closely follows the development of glomerular filtration, while digoxin clearance is, in part, affected by the maturation of tubular secretion capacity [10][11]. These PK profiles are further affected by a diverse set of nonmaturational covariates, including but not limited to genetics, disease characteristics (e.g., obesity, chronic inflammation, critical illness, chronic kidney disease, asphyxia) or environmental factors (e.g., type of nutrition, comedication, drug formulation, treatment modalities like therapeutic hypothermia) [12].
In pediatric pharmacotherapy, it should not be taken for granted that a given level of drug exposure to adults will result in similar drug effects in children, as concentration-effect profiles may also display developmental PD [13]. This refers to the ontogeny of biologic systems, and how drug (side-) effects are determined by developmental stage. Safety PD outcome parameters of specific interest relate to neurodevelopmental outcome, growth (length, specific organs) or pubertal development. Differences in chloride flow direction following activation of the GABA receptor, the maturational effect of cyclosporine on monocyte proliferation or the maturational QTc prolongation serve as illustrations of the critical relevance of maturational PD [14,15,16].
In pediatric pharmacotherapy, it should not be taken for granted that a given level of drug exposure to adults will result in similar drug effects in children, as concentration-effect profiles may also display developmental PD [13]. This refers to the ontogeny of biologic systems, and how drug (side-) effects are determined by developmental stage. Safety PD outcome parameters of specific interest relate to neurodevelopmental outcome, growth (length, specific organs) or pubertal development. Differences in chloride flow direction following activation of the GABA receptor, the maturational effect of cyclosporine on monocyte proliferation or the maturational QTc prolongation serve as illustrations of the critical relevance of maturational PD [14][15][16].
It is critically important that all sources of information are leveraged to optimize dose selection for neonates, infants or children, taking their (patho)physiology and associated variability into account and covering both PK and PD. Such sources include data from previous studies in humans (adults, other pediatric subpopulations), but also nonclinical juvenile animal models, in vitro systems, and in silico models. Depending on the drug development program, each of these methodological dimensions can be used to varying degrees, considering its strengths and limitations.
Among nonclinical in vivo models, large animals, like the neonatal and juvenile pig, are of increasing interest in pediatric drug development from two perspectives. First, to investigate and consequently better understand the mechanism of a disease, particularly when it is unique to pediatric patients. Second, the model may also provide important safety data for the pediatric population when performing juvenile toxicity studies. The choice of species and the design of juvenile toxicity studies are therefore the result of a series of complex considerations, including the therapeutic use of the drug, the age at which children will be treated, the duration of treatment, and potential age- or species-specific differences in efficacy, PK, or toxicity observed in adult animals [17]. The utility of a ‘leverage concept’ for dose determination and drug development programs in neonates has recently been described in this journal [18]. The following scenarios can be distinguished:
Pediatric disease similar to that in adults and/or older pediatric patients where dosing is known for adult and/or older pediatric patients = extrapolation of efficacy from adults to pediatric patients is permitted, and even supported [19,20].
Pediatric disease similar to that in adults and/or older pediatric patients where dosing is known for adult and/or older pediatric patients = extrapolation of efficacy from adults to pediatric patients is permitted, and even supported [19][20].
Pediatric disease related but not similar to that in adults and/or older pediatric patients where dosing is known for adult and/or older pediatric patients = additional information can be leveraged from either in vitro or in vivo models to guide initial dosing [19,20].
Pediatric disease related but not similar to that in adults and/or older pediatric patients where dosing is known for adult and/or older pediatric patients = additional information can be leveraged from either in vitro or in vivo models to guide initial dosing [19][20].
Pediatric disease unique to a given (sub)population within pediatrics, where these drugs are not utilized for these specific diseases in adults [19,20].
Pediatric disease unique to a given (sub)population within pediatrics, where these drugs are not utilized for these specific diseases in adults [19][20].
Even in the setting of similarity, additional research in juvenile animals may still be warranted when concerns related to developmental toxicology (like growth, neurodevelopment, kidney or cardiovascular system) should be addressed. Only about 10% of the 400 products (almost exclusive new drug approvals) of which the labels were reviewed between 1998–2009 by the FDA contained information on juvenile animals [21]. In a recent survey on European Pediatric Investigation Plan (PIP) decisions (2007–2017, 229 drugs) with juvenile animal requests, general toxicological studies were the most applicable study designs, with infectious diseases, endocrinology, neurology and cardiovascular diseases being the most common therapeutic areas. As anticipated, about 80% of these studies were in rats, while studies in pigs were limited (4.2%) [22]. Interestingly, a recent European Medicines Agency (EMA) analysis on juvenile animal studies in the field of anticancer drug research documented that juvenile models also generated evidence regarding new target organ toxicity (kidney, central and peripheral nervous system, impaired learning or memory, cardiac system) or increased severity of toxicity (including mortality rate) [23]. At the other end of the spectrum, with diseases that are unique to a given subpopulation within pediatrics, pig models can be instrumental in drug discovery and development for accurate mechanistic understanding of the disease or condition. Specific to neonates, this has been described for, e.g., necrotizing enterocolitis (NEC), resuscitation practices, or asphyxia. Studies in pigs have established the essential roles of prematurity, microbial colonization and enteral nutrition in the pathogenesis of NEC [24]. The (juvenile) pig is also an important animal model in research on human resuscitation [25]. In addition, in vivo data generated in neonatal animals—including (mini)pig—facilitate the development of a neonatal physiology-based PK (PBPK) model during therapeutic hypothermia [26].
2. Anatomical, Physiological and Developmental Similarities between Pigs and Humans
2.1. Characterization of the Pig As a Relevant Animal Model
From a drug discovery and development point of view, both mechanistic understanding and safety are key aspects in the process. Animal models used in the drug discovery process usually share pathophysiological traits with humans, which facilitates the identification of molecular targets and PD parameters. On the other hand, similar ADME process are desirable when the model is to be used in safety studies. Among nonrodent species, the pig presents several advantages related to similarities in anatomy and physiology when compared to humans that have been extensively reported and reviewed in literature [27,28,29,30,31]. Besides species similarity, the large litter size allows for a reduction in animals to be kept for breeding purposes, further facilitated by the short reproductive cycle [32]. Moreover, this enables researchers to place siblings into different experimental groups. Finally, large litter sizes usually lead to the spontaneous birth of intrauterine growth restricted (IUGR) pigs [33], which will be discussed later in this review. Pigs, especially minipigs (for size reasons), are relatively easy to handle and train [34,35], and their larger size at birth when compared to dogs or nonhuman primates (NHP) facilitates sampling at early stages. This larger size at birth, more similar to human neonates, facilitates the adaptation of NICU equipment for its use in pigs, increasing their translational value. Moreover, all routes of administration are possible and they represent the best model for dermal studies [36]. Pigs are precocial animals, which allows early separation from the mother. This can be beneficial in studies that require artificial rearing. Some disadvantages that will be further discussed in Section 4 include the more limited historical control data available for (mini)pigs than dogs and NHPs at present, the different placentation and a relatively more mature respiratory and musculoskeletal systems compared to human newborns. This is nevertheless compensated for by the easy access to spontaneous IUGR and preterm pigs showing different degrees of immaturity in several organs (see Section 3.2.2). These features have made the pig a well-accepted translational model, and in recent decades, several small to very small pig breeds have been developed specifically for laboratory use. For a detailed overview of the development and use of miniature, micro- and mini- pigs in biomedical research, we refer to Swindle et al., 2012 [29]. Within Europe, one of the most commonly used, purpose-bred pigs is the Göttingen Minipig. It is the result of crossbreeding the Minnesota Minipig, having a small stature and a gentle temperament, the Vietnamese potbelly pig with low body weight and high fertility, and the German Landrace pig for its white skin. Due to the fact that it is a genetically coherent breed that is easy to handle and can be housed in facilities originally designed for Beagle dogs, it is a popular model for drug development programs. Besides purpose-bred pigs, the wide variety of breeds used for pork production provides researchers with a broad spectrum of swine varieties, with different genetic backgrounds, sizes and fattening levels.
From a drug discovery and development point of view, both mechanistic understanding and safety are key aspects in the process. Animal models used in the drug discovery process usually share pathophysiological traits with humans, which facilitates the identification of molecular targets and PD parameters. On the other hand, similar ADME process are desirable when the model is to be used in safety studies. Among nonrodent species, the pig presents several advantages related to similarities in anatomy and physiology when compared to humans that have been extensively reported and reviewed in literature [27][28][29][30][31]. Besides species similarity, the large litter size allows for a reduction in animals to be kept for breeding purposes, further facilitated by the short reproductive cycle [32]. Moreover, this enables researchers to place siblings into different experimental groups. Finally, large litter sizes usually lead to the spontaneous birth of intrauterine growth restricted (IUGR) pigs [33], which will be discussed later in this review. Pigs, especially minipigs (for size reasons), are relatively easy to handle and train [34][35], and their larger size at birth when compared to dogs or nonhuman primates (NHP) facilitates sampling at early stages. This larger size at birth, more similar to human neonates, facilitates the adaptation of NICU equipment for its use in pigs, increasing their translational value. Moreover, all routes of administration are possible and they represent the best model for dermal studies [36]. Pigs are precocial animals, which allows early separation from the mother. This can be beneficial in studies that require artificial rearing. This is nevertheless compensated for by the easy access to spontaneous IUGR and preterm pigs showing different degrees of immaturity in several organs. These features have made the pig a well-accepted translational model, and in recent decades, several small to very small pig breeds have been developed specifically for laboratory use. For a detailed overview of the development and use of miniature, micro- and mini- pigs in biomedical research, we refer to Swindle et al., 2012 [29]. Within Europe, one of the most commonly used, purpose-bred pigs is the Göttingen Minipig. It is the result of crossbreeding the Minnesota Minipig, having a small stature and a gentle temperament, the Vietnamese potbelly pig with low body weight and high fertility, and the German Landrace pig for its white skin. Due to the fact that it is a genetically coherent breed that is easy to handle and can be housed in facilities originally designed for Beagle dogs, it is a popular model for drug development programs. Besides purpose-bred pigs, the wide variety of breeds used for pork production provides researchers with a broad spectrum of swine varieties, with different genetic backgrounds, sizes and fattening levels.
Although pigs have been models in biomedical research for decades, efforts are still being made to better characterize this model. As an example, the COST action called SALAAM (Sharing Advances on Large Animal Models), a EU-funded research network (2014–2018), connected researchers from 24 European countries with the objective of improving large animal models and phenotyping protocols, developing selection criteria for animal models and creating and sharing data and samples to advance the use of large animals (pigs, small ruminants and rabbits) where they may be of interest. However, despite already having in-depth knowledge on the anatomy and physiology of the (mini)pig and attempts to further characterize ADME processes in minipigs for their use in pharmaceutical research [32,37,38,39], more work is needed, especially in neonatal and juvenile pigs.
Although pigs have been models in biomedical research for decades, efforts are still being made to better characterize this model. As an example, the COST action called SALAAM (Sharing Advances on Large Animal Models), a EU-funded research network (2014–2018), connected researchers from 24 European countries with the objective of improving large animal models and phenotyping protocols, developing selection criteria for animal models and creating and sharing data and samples to advance the use of large animals (pigs, small ruminants and rabbits) where they may be of interest. However, despite already having in-depth knowledge on the anatomy and physiology of the (mini)pig and attempts to further characterize ADME processes in minipigs for their use in pharmaceutical research [32][37][38][39], more work is needed, especially in neonatal and juvenile pigs.
2.2. The Pig in Pediatric Research
2.2.1. Pig and Human Postnatal Development
In order to assess the feasibility of the neonatal and juvenile pig as models for pediatric drug development, an in-depth characterization of this model must be carried out in the first place, followed by a comparison with the anatomical, physiological and ADME characteristics in the corresponding pediatric age groups. Our group has already reported on the age-related maturation of organ weights in the developing Göttingen Minipig in an effort to further develop a PBPK model [35], but more data are needed. The implementation of this model would benefit from data on microsomal protein per gram of liver and abundance on drug metabolizing enzymes during development, or from a better understanding of pig orthologues for human cytochrome P450 (CYP) enzymes. For the developing domestic pig, the anatomy, physiology and the absorption, distribution and excretion of drugs have been reviewed by Gasthuys et al. [40]. As some of the above data are publicly accessible in the ICH S11 guidelines on nonclinical safety testing in support of the development of pediatric pharmaceuticals [36], we will only highlight some key points in this review. The EMA has established different age categories within the pediatric population [41], and many similarities between human and Göttingen Minipig organ development (as the reference breed used in the pharmaceutical industry [42] were reported in the ICH S11 guidelines [36].
In general, pigs and humans share many developmental milestones: The patterns of development of the gastrointestinal tract (GIT), the cardiovascular, the CNS systems and the eye are quite similar in both species, while renal, immune and reproductive development occur slightly earlier and more quickly in humans than in pigs. These data are illustrated in
.
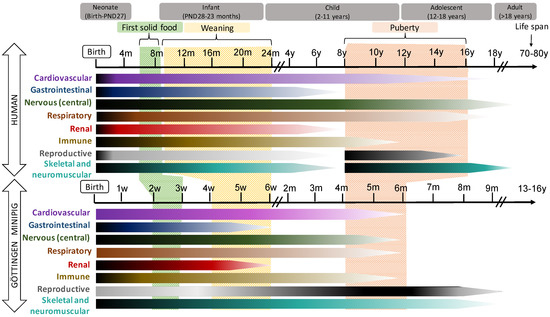
Figure 1.
Schematic representation of the postnatal development of different organ systems in human (top) and Göttingen Minipig. In the horizontal bars, the intensity of the maturation process is represented by dark (more intense) and light (less intense) tones. The time bar represents weeks (w), months (m) or year (y) of life.
2.2.2. Hepatic Drug Metabolism in the Neonatal and Juvenile Pig
Hepatic Phase I drug metabolism mediated by CYP enzymes has been investigated extensively in adult conventional pig strains [72,73,74,75,76,77,78,79,80,81] and minipig strains [43,44,79,82,83,84,85,86,87] over the past 30 years. Knowledge on the ontogeny of these processes in the neonatal and juvenile population is much more limited. Particularly in neonates, it is crucial to predict drug disposition correctly in order to avoid inefficacy due to underdosing or adverse effects caused by overdosing. Recently, CYP activity was determined in the neonatal and juvenile conventional pig [88] and the Göttingen Minipig [45] in different age groups using several human CYP450 substrates. As such, substrate specificity was examined and CYP450 activity levels in (mini)pig were compared to those in human. In the Göttingen Minipig, we found that CYP450 enzyme activity increased postnatally. However, differences in onset and speed in development were observed: CYP1A2- and CYP2D6-like activity levels increased fast during the first week of life, whereas CYP2C9- and CYP3A4-like activities matured more slowly, reaching their highest levels in 1-month-old pigs [45], corresponding roughly to a 2-year-old child [89,90,91,92,93]. In the conventional pig, similar results were obtained [88]. In addition, no sex-related differences were observed in the neonatal and juvenile age groups regarding the CYP450 ontogeny patterns until puberty [45,88]. With regard to CYP450 protein abundance, some research has already been conducted on the conventional pig [88], and this question is currently being addressed in the Göttingen Minipig by our group. In general, activity and abundance data correlate well, although CYP isoform-specific differences have been reported [88]. Regarding Phase 2 metabolism, data are scarce. A recent in vitro study on 1-day and 2-, 5-, 10- and 20-week old male Camborough-29 pigs showed that UDP Glucuronosyltransferase (UGT) enzyme activity increased from day 1 until week 10, followed by a decline at around week 20 [94]. An in vivo study with ibuprofen in 1-, 4-, and 8-week old, and 6–7-month old, mixed breed pigs also showed UGT activity already in neonatal pigs [95]. In our group, UGT activity was investigated in Göttingen Minipigs with age groups ranging from the late fetal stage until postnatal day 28, and adults [45]. From PND 7 onward, UGT activity increased without sex-related differences [45]. In the youngest age groups (gestational day age (GDA) 84–86, GDA 108, postnatal (PND) day 1 and 3), activities were below the lower detection limit when using a luminescence-based assay [45]. However, immunohistochemical analysis showed that even in the late fetal stages, UGT1A could be detected. In accordance with the activity results, UGT1A detection increased with age [45]. In general, it can be concluded that UGT enzymes are expressed from an early age, but further characterization of the different isoforms is needed in order to better predict drug disposition in this animal model. With regard to drug transport (often referred to as Phase 0 for uptake transporters and Phase III for efflux transporters), the data in the pig are even more scarce. For the Göttingen Minipig, we performed a semiquantitative assessment of P-glycoprotein (P-gp, encoded by the Multidrug Resistance Gene,
Hepatic Phase I drug metabolism mediated by CYP enzymes has been investigated extensively in adult conventional pig strains [72][73][74][75][76][77][78][79][80][81] and minipig strains [43][44][79][82][83][84][85][86][87] over the past 30 years. Knowledge on the ontogeny of these processes in the neonatal and juvenile population is much more limited. Particularly in neonates, it is crucial to predict drug disposition correctly in order to avoid inefficacy due to underdosing or adverse effects caused by overdosing. Recently, CYP activity was determined in the neonatal and juvenile conventional pig [88] and the Göttingen Minipig [45] in different age groups using several human CYP450 substrates. As such, substrate specificity was examined and CYP450 activity levels in (mini)pig were compared to those in human. In the Göttingen Minipig, we found that CYP450 enzyme activity increased postnatally. However, differences in onset and speed in development were observed: CYP1A2- and CYP2D6-like activity levels increased fast during the first week of life, whereas CYP2C9- and CYP3A4-like activities matured more slowly, reaching their highest levels in 1-month-old pigs [45], corresponding roughly to a 2-year-old child [89][90][91][92][93]. In the conventional pig, similar results were obtained [88]. In addition, no sex-related differences were observed in the neonatal and juvenile age groups regarding the CYP450 ontogeny patterns until puberty [45][88]. With regard to CYP450 protein abundance, some research has already been conducted on the conventional pig [88], and this question is currently being addressed in the Göttingen Minipig by our group. In general, activity and abundance data correlate well, although CYP isoform-specific differences have been reported [88]. Regarding Phase 2 metabolism, data are scarce. A recent in vitro study on 1-day and 2-, 5-, 10- and 20-week old male Camborough-29 pigs showed that UDP Glucuronosyltransferase (UGT) enzyme activity increased from day 1 until week 10, followed by a decline at around week 20 [94]. An in vivo study with ibuprofen in 1-, 4-, and 8-week old, and 6–7-month old, mixed breed pigs also showed UGT activity already in neonatal pigs [95]. In our group, UGT activity was investigated in Göttingen Minipigs with age groups ranging from the late fetal stage until postnatal day 28, and adults [45]. From PND 7 onward, UGT activity increased without sex-related differences [45]. In the youngest age groups (gestational day age (GDA) 84–86, GDA 108, postnatal (PND) day 1 and 3), activities were below the lower detection limit when using a luminescence-based assay [45]. However, immunohistochemical analysis showed that even in the late fetal stages, UGT1A could be detected. In accordance with the activity results, UGT1A detection increased with age [45]. In general, it can be concluded that UGT enzymes are expressed from an early age, but further characterization of the different isoforms is needed in order to better predict drug disposition in this animal model. With regard to drug transport (often referred to as Phase 0 for uptake transporters and Phase III for efflux transporters), the data in the pig are even more scarce. For the Göttingen Minipig, we performed a semiquantitative assessment of P-glycoprotein (P-gp, encoded by the Multidrug Resistance Gene,
MDR
) in the liver of neonatal and juvenile pigs and fetuses using immunohistochemistry. No difference was observed in P-gp expression between livers from GDA84 to adult animals (1.5–3 years of age) [43].
When comparing the above data with the ontogeny profile of the drug disposition processes in human, which have been reviewed extensively elsewhere [96,97,98], remarkable similarities are present. For UGT and P-gp, the ontogeny profile on protein and activity level, if assessed, is very similar. With regard to the CYP activity, the interpretation is more complex. The slow maturation profile of CYP2C9 and CYP3A4 activity in (mini)pigs corresponds well with the pediatric population. For CYP1A2 and CYP2D6, there appears to be an earlier onset of activity in the pig than in human, and CYP2D6 activity in general appears to be much higher than in human. Still, when comparing the pig with man, one needs to be very cautious, as studies may use different substrates or other testing conditions, which may confound the results and, as such, species comparisons. This said, even when not directly translatable to human, in vitro and in vivo drug metabolism data in juvenile animals are critical, as they may explain differences in efficacy or toxicity with the human population, and they can be used in mathematical models to better predict exposure, especially in the very young age groups, as further discussed in the next section.
When comparing the above data with the ontogeny profile of the drug disposition processes in human, which have been reviewed extensively elsewhere [96][97][98], remarkable similarities are present. For UGT and P-gp, the ontogeny profile on protein and activity level, if assessed, is very similar. With regard to the CYP activity, the interpretation is more complex. The slow maturation profile of CYP2C9 and CYP3A4 activity in (mini)pigs corresponds well with the pediatric population. For CYP1A2 and CYP2D6, there appears to be an earlier onset of activity in the pig than in human, and CYP2D6 activity in general appears to be much higher than in human. Still, when comparing the pig with man, one needs to be very cautious, as studies may use different substrates or other testing conditions, which may confound the results and, as such, species comparisons. This said, even when not directly translatable to human, in vitro and in vivo drug metabolism data in juvenile animals are critical, as they may explain differences in efficacy or toxicity with the human population, and they can be used in mathematical models to better predict exposure, especially in the very young age groups, as further discussed in the next section.
2.2.3. PBPK Models in the Neonatal and Juvenile Pig
In general, PBPK models provide a mechanistic framework for predicting drug exposure in special populations via a ‘full bottom-up’ modelling strategy. Input data include: (i) drug-specific data, including physicochemical properties and in vitro disposition data (e.g., metabolic rates, plasma protein binding and transepithelial permeability; and (ii) quantitative mapping of the physiology of the biologic system of interest (e.g., neonatal human or pig). The fact that predictions of in vivo drug disposition can be made based on ‘first principles’, i.e., with limited need for in vivo animal or clinical data remains one of the unique strengths of PBPK models. However, at present, this important advantage also comes at a cost, i.e., (i) predicting population variability in drug disposition processes remains challenging; (ii) initial evaluation of the predictive performance is difficult in populations (e.g., neonates) where limited clinical data have been collected, especially for first-in-class compounds.
When no or limited clinical data are available for model verification, building a PBPK model for a corresponding animal model carries the potential to indirectly inform (and improve) the desired PBPK model in the human target population. This strategy has, for instance, been applied to determine an adequate IV dosing regimen for oseltamivir in neonates and infants. In this case, the availability of plasma and liver PK data obtained after oral and IV administration in newborn and adult marmosets was instrumental in establishing and verifying neonate/adult PBPK models for oseltamivir in this species. Moreover, confidence in the ultimate human neonatal PBPK models was achieved by input from both the newborn marmoset PBPK model (in terms of immaturity) and from the adult human PBPK model (more in terms of incorporation of drug-specific data).
It is anticipated that also in (neonatal and juvenile) pigs, PBPK models will be increasingly used to enhance the chance for successful development of PBPK models in equivalent human populations. Consistently, best practice guidelines for building PBPK models for ‘novel’ species (such as minipig) have very recently been proposed [99]. Specifically, for supporting the development of (mini)pig PBPK models, it is critical that the relevant physiological descriptors are available in sufficient detail. For the adult Göttingen Minipig, Suenderhauf and Parrott [39] were the first to publish a compilation of gastrointestinal pH values and transit times, along with organ sizes and blood perfusion rates. The Advanced Compartmental and Transit (ACAT) concept was used to describe the drug absorption processes. The PBPK model implementation of these physiological data was verified with moxifloxaxin and griseofulvin, both after IV and oral administration. In a follow-up study [100], the same research group explored gastric emptying times (GET) in minipigs using paracetamol as a model drug. Their findings demonstrated high variability in GET values that also turned out to be higher as compared to humans. Another absorption-related application of PBPK modelling included the development of modified release formulation for a compound with region-dependent absorption [101]. Furthermore, using both slowly eliminated and rapidly cleared model compounds, the utility of PBPK modelling to extrapolate PK profiles in minipigs to the human situation has been illustrated [102]. More recently, predictions of the volume of distribution in minipigs (Vd,ss) based on tissue compositions (e.g., relative amounts of neutral lipids, phospholipids and intra/extracellular water) have been implemented, thereby making a major step in the further refinement of a Göttingen Minipig PBPK concept [103]. To support establishing minipig PBPK models for the younger age groups, we previously reported organ weights and GI pH values of Göttingen Minipigs between the fetal stage and 5 months of age as a first step [35]. Several critical data, such as liver blood flow, abundance and activity of drug transporters, scaling factors, etc. are still missing, but with the increasing availability of ontogeny data for both phase I and phase II biotransformation pathways, the pieces of the puzzle to build a neonatal Göttingen Minipig PBPK model are falling into place.
Ultimately, development of PBPK models for human neonates with a specific disease is also expected to benefit from the learnings obtained when developing PBPK models in the corresponding neonatal animal model. As recently proposed by our group [26], the goal to establish both a Göttingen Minipig and human neonatal PBPK model is expected to prove uniquely instrumental in predicting the influence of therapeutic hypothermia on PK of key drugs used in asphyxiated neonates. The advantage of incorporating a PBPK platform in such an endeavour lays in the possibility to deconvolute the distinct influences of disease severity, therapeutic options and maturational physiology on PK. Importantly, this latter project will also require tailored in vitro studies—for instance, to study the influence of temperature on intrinsic enzyme-mediated clearance—to be used as input data for the PBPK models. For the purpose of this project, the proposed workflow for the development of PBPK models will be as follows: (i) build healthy neonatal pig PBPK model; (ii) modify the PBPK model to reflect hypotheses regarding the impact of asphyxia and/or hypothermia on physiology and PK pathways; (iii) iterative evaluation and improvement of PBPK-based predictions against PK observations obtained in neonatal pigs; (iv) establish a healthy neonatal human PBPK model; (v) build a human neonatal PBPK model for asphyxiated neonates, implementing the verified hypotheses regarding disease impact as evaluated in the corresponding Göttingen Minipig disease model.
References
- Anker, J.V.D.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25.
- Germovsek, E.; Barker, C.I.S.; Sharland, M.; Standing, J.F. Scaling clearance in paediatric pharmacokinetics: All models are wrong, which are useful? Br. J. Clin. Pharmacol. 2017, 83, 777–790.
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Gesquiere, I.; Greupink, R.; Keszthelyi, D.; Koskinen, M.; et al. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280.
- Van Groen, B.D.; Krekels, E.H.J.; Mooij, M.G.; Van Duijn, E.; Vaes, W.H.J.; Windhorst, A.D.; Van Rosmalen, J.; Hartman, S.J.F.; Hendrikse, N.H.; Koch, B.C.P.; et al. The Oral Bioavailability and Metabolism of Midazolam in Stable Critically Ill Children: A Pharmacokinetic Microtracing Study. Clin. Pharmacol. Ther. 2021, 109, 140–149.
- Ward, R.M.; the International Neonatal Consortium (INC); Benjamin, D.; Barrett, J.S.; Allegaert, K.; Portman, R.; Davis, J.M.; Turner, M.A. Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr. Res. 2016, 81, 692–711.
- Oyaert, M.; Spriet, I.; Allegaert, K.; Smits, A.; Vanstraelen, K.; Peersman, N.; Wauters, J.; Verhaegen, J.; Vermeersch, P.; Pauwels, S. Factors Impacting Unbound Vancomycin Concentrations in Different Patient Populations. Antimicrob. Agents Chemother. 2015, 59, 7073–7079.
- Allegaert, K.; Anker, J.V.D. Ontogeny of Phase I Metabolism of Drugs. J. Clin. Pharmacol. 2019, 59, S33–S41.
- Cristea, S.; Krekels, E.H.J.; Rostami-Hodjegan, A.; Allegaert, K.; Knibbe, C.A.J. The Influence of Drug Properties and Ontogeny of Transporters on Pediatric Renal Clearance through Glomerular Filtration and Active Secretion: A Simulation-Based Study. AAPS J. 2020, 22, 1–10.
- Cheung, K.W.K.; Van Groen, B.D.; Spaans, E.; Van Borselen, M.D.; De Bruijn, A.C.; Simons-Oosterhuis, Y.; Tibboel, D.; Samsom, J.N.; Verdijk, R.M.; Smeets, B.; et al. A Comprehensive Analysis of Ontogeny of Renal Drug Transporters: mRNA Analyses, Quantitative Proteomics, and Localization. Clin. Pharmacol. Ther. 2019, 106, 1083–1092.
- De Cock, R.F.W.; Allegaert, K.; Brussee, J.M.; Sherwin, C.M.T.; Mulla, H.; De Hoog, M.; Anker, J.N.V.D.; Danhof, M.; Knibbe, C.A.J. Simultaneous Pharmacokinetic Modeling of Gentamicin, Tobramycin and Vancomycin Clearance from Neonates to Adults: Towards a Semi-physiological Function for Maturation in Glomerular Filtration. Pharm. Res. 2014, 31, 2643–2654.
- Aladjem, M.; Kaplinsky, C.; Wolfish, N.; Laufer, Y.; Halkin, H. Maturation of Renal Tubular Transport of Digoxin. Pediatr. Res. 1981, 15, 282–283.
- Allegaert, K.; Simons, S.; Tibboel, D.; Krekels, E.H.; Knibbe, C.A.; Anker, J.N.V.D. Non-maturational covariates for dynamic systems pharmacology models in neonates, infants, and children: Filling the gaps beyond developmental pharmacology. Eur. J. Pharm. Sci. 2017, 109, S27–S31.
- Mulla, H. Understanding Developmental Pharmacodynamics. Pediatr. Drugs 2010, 12, 223–233.
- Rakhade, S.N.; Jensen, F.E. Epileptogenesis in the immature brain: Emerging mechanisms. Nat. Rev. Neurol. 2009, 5, 380–391.
- Marshall, J.A.; Kearns, G.L. Developmental pharmacodynamics of cyclosporine. Clin. Pharmacol. Ther. 1999, 66, 66–75.
- Krasemann, T.; Bente, K.; Burkhardtsmaier, G. The corrected QT interval in 24 h ECGs in neonates. Clin. Res. Cardiol. 2010, 99, 309–314.
- Barrow, P.; Schmitt, G. Juvenile Nonclinical Safety Studies in Support of Pediatric Drug Development. Adv. Struct. Saf. Stud. 2017, 1641, 25–67.
- Anker, J.V.D.; McCune, S.; Annaert, P.; Baer, G.R.; Mulugeta, Y.; Abdelrahman, R.; Wu, K.; Krudys, K.M.; Fisher, J.; Slikker, W.; et al. Approaches to Dose Finding in Neonates, Illustrating the Variability between Neonatal Drug Development Programs. Pharmaceutics 2020, 12, 685.
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/paediatric-medicines/paediatric-regulation (accessed on 23 October 2020).
- Food and Drug Administration. Draft Guidance: Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products. Guidance for Industry. Available online: https://www.fda.gov/media/133660/download (accessed on 23 October 2020).
- Tassinari, M.S.; Benson, K.; Elayan, I.; Espandiari, P.; Davis-Bruno, K. Juvenile animal studies and pediatric drug development retrospective review: Use in regulatory decisions and labeling. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2011, 92, 261–265.
- Baldrick, P. Juvenile Animal Testing: Assessing Need and Use in the Drug Product Label. Ther. Innov. Regul. Sci. 2018, 52, 641–648.
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/results-juvenile-animal-studies-jas-impact-anti-cancer-medicine-development-use-children_en.pdf (accessed on 23 October 2020).
- Burrin, D.; Sangild, P.T.; Stoll, B.; Thymann, T.; Buddington, R.; Marini, J.; Olutoye, O.; Shulman, R.J. Translational Advances in Pediatric Nutrition and Gastroenterology: New Insights from Pig Models. Annu. Rev. Anim. Biosci. 2020, 8, 321–354.
- Barouxis, D.; Chalkias, A.; Syggelou, A.; Iacovidou, N.; Xanthos, T. Research in human resuscitation: What we learn from animals. J. Matern. Neonatal Med. 2012, 25, 44–46.
- Smits, A.; Annaert, P.; Van Cruchten, S.; Allegaert, K. A Physiology-Based Pharmacokinetic Framework to Support Drug Development and Dose Precision During Therapeutic Hypothermia in Neonates. Front. Pharmacol. 2020, 11.
- Sciascia, Q.; Das, G.; Metges, C.C. Review: The pig as a model for humans: Effects of nutritional factors on intestinal function and health1. J. Anim. Sci. 2016, 94, 441–452.
- Mudd, A.T.; Dilger, R. Early-Life Nutrition and Neurodevelopment: Use of the Piglet as a Translational Model. Adv. Nutr. 2017, 8, 92–104.
- Swindle, M.; Makin, A.; Herron, A.; Clubb, F., Jr.; Frazier, K. Swine as models in biomedical research and toxicology testing. Vet. Pathol. 2012, 49, 344–356.
- Swindle, M.M.; Smith, A.C. Swine in the Laboratory: Surgery, Anesthesia, Imaging, and Experimental Techniques; CRC Press: Boca Raton, FL, USA, 2015.
- Butler, J.E.; Sun, J.; Wertz, N.; Sinkora, M. Antibody repertoire development in swine. Dev. Comp. Immunol. 2006, 30, 199–221.
- Forster, R.; Bode, G.; Ellegaard, L.; Van Der Laan, J.W. The RETHINK project on minipigs in the toxicity testing of new medicines and chemicals: Conclusions and recommendations. J. Pharmacol. Toxicol. Methods 2010, 62, 236–242.
- Ferenc, K.; Pietrzak, P.; Godlewski, M.M.; Piwowarski, J.; Kilianczyk, R.; Guilloteau, P.; Zabielski, R. Intrauterine growth retarded piglet as a model for humans-Studies on the perinatal development of the gut structure and function. Reprod. Biol. 2014, 14, 51–60.
- Stricker-Krongrad, A.; Shoemake, C.R.; Bouchard, G.F. The Miniature Swine as a Model in Experimental and Translational Medicine. Toxicol. Pathol. 2016, 44, 612–623.
- Van Peer, E.; Downes, N.; Casteleyn, C.; Van Ginneken, C.; Weeren, A.; Van Cruchten, S. Organ data from the developing Göttingen minipig: First steps towards a juvenile PBPK model. J. Pharmacokinet. Pharmacodyn. 2016, 43, 179–190.
- ICH. Ich Guideline s11 on Nonclinical Safety Testing in Support of Development of Paediatric Pharmaceuticals; Committee for Medicinal Products for Human Use, 2020; Available online: https://www.ema.europa.eu/en/ich-guideline-s11-nonclinical-safety-testing-support-development-paediatric-pharmaceuticals-step-5vv (accessed on 18 October 2020).
- Summerfield, A.; Rziha, H.-J.; Saalmüller, A. Functional characterization of porcine CD4+ CD8+ extrathymic T lymphocytes. Cell. Immunol. 1996, 168, 291–296.
- Svendsen, O. (Ed.) The minipig in toxicology. In Proceedings of the Satellite Symposium to Eurotox’97, Aarhus, Denmark; 1997.
- Suenderhauf, C.; Parrott, N. A Physiologically Based Pharmacokinetic Model of the Minipig: Data Compilation and Model Implementation. Pharm. Res. 2012, 30, 1–15.
- Gasthuys, E.; Vandecasteele, T.; De Bruyne, P.; Walle, J.V.; De Backer, P.; Cornillie, P.; Devreese, M.; Croubels, S. The potential use of piglets as human pediatric surrogate for preclinical pharmacokinetic and pharmacodynamic drug testing. Curr. Pharm. Des. 2016, 22, 4069–4085.
- Baber, N. Guide to Paediatric Clinical Research. Br. J. Clin. Pharmacol. 2008, 65, 282.
- Heining, P.; Ruysschaert, T. The Use of Minipig in Drug Discovery and Development. Toxicol. Pathol. 2016, 44, 467–473.
- Van Peer, E.M.; Verbueken, E.; Saad, M.; Casteleyn, C.; Van Ginneken, C.J.; Van Cruchten, S. Ontogeny of CYP3A and P-Glycoprotein in the Liver and the Small Intestine of the Göttingen Minipig: An Immunohistochemical Evaluation. Basic Clin. Pharmacol. Toxicol. 2013, 114, 387–394.
- Van Peer, E.M.; De Bock, L.; Boussery, K.; Van Bocxlaer, J.; Casteleyn, C.; Van Ginneken, C.; Van Cruchten, S. Age-related Differences in CYP3A Abundance and Activity in the Liver of the Göttingen Minipig. Basic Clin. Pharmacol. Toxicol. 2015, 117, 350–357.
- Van Peer, E.; Jacobs, F.; Snoeys, J.; Van Houdt, J.; Pijpers, I.; Casteleyn, C.; Van Ginneken, C.; Van Cruchten, S. In vitro Phase I-and Phase II-Drug Metabolism in The Liver of Juvenile and Adult Göttingen Minipigs. Pharm. Res. 2017, 34, 750–764.
- Crick, S.J.; Sheppard, M.N.; Ho, S.Y.; Gebstein, L.; Anderson, R.H. Anatomy of the pig heart: Comparisons with normal human cardiac structure. J. Anat. 1998, 193, 105–119.
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83.
- Butler, J.E.; Lager, K.; Splichal, I.; Francis, D.; Kacskovics, I.; Sinkora, M.; Wertz, N.; Sun, J.; Zhao, Y.; Brown, W.; et al. The piglet as a model for B cell and immune system development. Veter-Immunol. Immunopathol. 2009, 128, 147–170.
- Comstock, S.S.; Reznikov, E.A.; Contractor, N.; Donovan, S.M. Dietary Bovine Lactoferrin Alters Mucosal and Systemic Immune Cell Responses in Neonatal Piglets. J. Nutr. 2014, 144, 525–532.
- Valent, D.; Yeste, N.; Hernández-Castellano, L.E.; Arroyo, L.; Wu, W.; García-Contreras, C.; Vazquez-Gomez, M.; González-Bulnes, A.; Bendixen, E.; Bassols, A. SWATH-MS quantitative proteomic investigation of intrauterine growth restriction in a porcine model reveals sex differences in hippocampus development. J. Proteom. 2019, 204, 103391.
- Gortner, L.; Shen, J.; Tutdibi, E. Sexual Dimorphism of Neonatal Lung Development. Klin. Pädiatrie 2013, 225, 64–69.
- Óvilo, C.; Gonzalez-Bulnes, A.; Benítez, R.; Ayuso, M.; Barbero, A.; Pérez-Solana, M.L.; Barragán, C.; Astiz, S.; Fernández, A.; López-Bote, C. Prenatal programming in an obese swine model: Sex-related effects of maternal energy restriction on morphology, metabolism and hypothalamic gene expression. Br. J. Nutr. 2014, 111, 735–746.
- Anadkat, J.S.; Kuzniewicz, M.W.; Chaudhari, B.P.; Cole, F.S.; Hamvas, A. Increased risk for respiratory distress among white, male, late preterm and term infants. J. Perinatol. 2012, 32, 780–785.
- Spengler, D.; Rintz, N.; Krause, M.F. An Unsettled Promise: The Newborn Piglet Model of Neonatal Acute Respiratory Distress Syndrome (NARDS). Physiologic Data and Systematic Review. Front. Physiol. 2019, 10.
- Ziegler, A.L.; Gonzalez, L.; Blikslager, A.T. Large Animal Models: The Key to Translational Discovery in Digestive Disease Research. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 716–724.
- Neal-Kluever, A.; Fisher, J.; Grylack, L.; Kakiuchi-Kiyota, S.; Halpern, W.G. Physiology of the Neonatal Gastrointestinal System Relevant to the Disposition of Orally Administered Medications. Drug Metab. Dispos. 2018, 47, 296–313.
- Fallingborg, J.; Christensen, L.A.; Ingeman-Nielsen, M.; Jacobsen, B.A.; Abildgaard, K.; Rasmussen, H.H.; Rasmussen, S.N. Measurement of Gastrointestinal pH and Regional Transit Times in Normal Children. J. Pediatr. Gastroenterol. Nutr. 1990, 11, 211–214.
- Bartelink, I.H.; Rademaker, C.M.A.; Schobben, A.F.A.M.; Anker, J.N.V.D. Guidelines on Paediatric Dosing on the Basis of Developmental Physiology and Pharmacokinetic Considerations. Clin. Pharmacokinet. 2006, 45, 1077–1097.
- Tan, A.Y.; Sourial, M.; Hutson, J.M.; Southwell, B.R. Short-Term Interferential Transabdominal Electrical Stimulation Did Not Change Oral-Rectal Transit Time in Piglets. Neuromodul. Technol. Neural Interface 2018, 21, 669–675.
- Skrzypek, T.; Piedra, J.V.; Skrzypek, H.; Kazimierczak, W.; Szymanczyk, S.; Zabielski, R. Changes in pig small intestinal absorptive area during the first 14days of life. Livest. Sci. 2010, 133, 53–56.
- Heinritz, S.N.; Mosenthin, R.; Weiss, E. Use of pigs as a potential model for research into dietary modulation of the human gut microbiota. Nutr. Res. Rev. 2013, 26, 191–209.
- Kim, M.Y.; Eiby, Y.A.; Lumbers, E.R.; Wright, L.L.; Gibson, K.J.; Barnett, A.C.; Lingwood, B.E. Effects of Glucocorticoid Exposure on Growth and Structural Maturation of the Heart of the Preterm Piglet. PLoS ONE 2014, 9, e93407.
- Christoffersen, B.Ø.; Jensen, S.J.; Ludvigsen, T.P.; Nilsson, S.K.; Grossi, A.B.; Heegaard, P.M.H. Age- and Sex-Associated Effects on Acute-Phase Proteins in Göttingen Minipigs. Comp. Med. 2015, 65, 333–341.
- Friis, C. Postnatal development of the pig kidney: Ultrastucure of the glomerulus and the proximal tubule. J. Anat. 1980, 130, 513–526.
- De Cock, R.F.; Allegaert, K.; Schreuder, M.F.; Sherwin, C.M.; de Hoog, M.; van den Anker, J.N.; Danhof, M.; Knibbe, C.A.J. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin. Pharmacokinet. 2012, 51, 105–117.
- Dhondt, L.; Croubels, S.; De Paepe, P.; Wallis, S.C.; Pandey, S.; Roberts, J.A.; Lipman, J.; De Cock, P.; Devreese, M. Conventional Pig as Animal Model for Human Renal Drug Excretion Processes: Unravelling the Porcine Renal Function by Use of a Cocktail of Exogenous Markers. Front. Pharmacol. 2020, 11.
- Dawson, H. A Comparative Assessment of the Pig, Mouse and Human Genomes; CRC Press: Boca Raton, FL, USA, 2011; pp. 323–342.
- Palermo, S.; Capra, E.; Torremorell, M.; Dolzan, M.; Davoli, R.; Haley, C.; Giuffra, E. Toll-like receptor 4genetic diversity among pig populations. Anim. Genet. 2009, 40, 289–299.
- Hole, C.V.; Goyens, J.; Prims, S.; Fransen, E.; Hernando, M.A.; Van Cruchten, S.; Aerts, P.; Van Ginneken, C. How innate is locomotion in precocial animals? A study on the early development of spatio-temporal gait variables and gait symmetry in piglets. J. Exp. Biol. 2017, 220, 2706–2716.
- Judge, E.P.; Hughes, J.M.L.; Egan, J.J.; Maguire, M.; Molloy, E.L.; O’Dea, S. Anatomy and Bronchoscopy of the Porcine Lung. A Model for Translational Respiratory Medicine. Am. J. Respir. Cell Mol. Biol. 2014, 51, 334–343.
- Haworth, S.G.; Hislop, A.A. Adaptation of the pulmonary circulation to extra-uterine life in the pig and its relevance to the human infant. Cardiovasc. Res. 1981, 15, 108–119.
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Cytochrome P450 Pig Liver Pie: Determination of Individual Cytochrome P450 Isoform Contents in Microsomes from Two Pig Livers Using Liquid Chromatography in Conjunction with Mass Spectrometry. Drug Metab. Dispos. 2011, 39, 2130–2134.
- Anzenbacherová, E.; Baranová, J.; Zuber, R.; Pěchová, A.; Anzenbacher, P.; Souček, P.; Martínková, J. Model Systems Based on Experimental Animals for Studies on Drug Metabolism in Man: (Mini)Pig Cytochromes P450 3A29 and 2E1. Basic Clin. Pharmacol. Toxicol. 2005, 96, 244–245.
- Brunius, C.; Rasmussen, M.K.; Lacoutière, H.; Andersson, K.; Ekstrand, B.; Zamaratskaia, G. Expression and activities of hepatic cytochrome P450 (CYP1A, CYP2A and CYP2E1) in entire and castrated male pigs. Animal 2012, 6, 271–277.
- Burkina, V.; Rasmussen, M.K.; Oliinychenko, Y.; Zamaratskaia, G. Porcine cytochrome 2A19 and 2E1. Basic Clin. Pharmacol. Toxicol. 2018, 124, 32–39.
- Kojima, M.; Degawa, M. Sex differences in constitutive mRNA levels of CYP2B22, CYP2C33, CYP2C49, CYP3A22, CYP3A29 and CYP3A46 in the pig liver: Comparison between Meishan and Landrace pigs. Drug Metab. Pharmacokinet. 2016, 31, 185–192.
- Schelstraete, W.; De Clerck, L.; Govaert, E.; Millecam, J.; Devreese, M.; Deforce, D.; Van Bocxlaer, J.; Croubels, S. Characterization of Porcine Hepatic and Intestinal Drug Metabolizing CYP450: Comparison with Human Orthologues from A Quantitative, Activity and Selectivity Perspective. Sci. Rep. 2019, 9, 1–14.
- Skaanild, M.T. Porcine cytochrome P450 and metabolism. Curr. Pharm. Des. 2006, 12, 1421–1427.
- Skaanild, M.T.; Friis, C. Cytochrome P450 Sex Differences in Minipigs and Conventional Pigs. Pharmacol. Toxicol. 1999, 85, 174–180.
- Skaanild, M.T.; Friis, C. Is Cytochrome P450 CYP2D Activity Present in Pig Liver? Pharmacol. Toxicol. 2002, 91, 198–203.
- Skaanild, M.T.; Friis, C. Analyses of CYP2C in Porcine Microsomes. Basic Clin. Pharmacol. Toxicol. 2008, 103, 487–492.
- Baranová, J.; Anzenbacherová, E.; Anzenbacher, P.; Souček, P. Minipig Cytochrome P450 2e1: Comparison with Human Enzyme. Drug Metab. Dispos. 2005, 33, 862–865.
- Heckel, T.; Schmucki, R.; Berrera, M.; Ringshandl, S.; Badi, L.; Steiner, G.; Ravon, M.; Küng, E.; Kuhn, B.; Kratochwil, N.A.; et al. Functional analysis and transcriptional output of the Göttingen minipig genome. BMC Genom. 2015, 16, 932.
- Lignet, F.; Sherbetjian, E.; Kratochwil, N.; Jones, R.; Suenderhauf, C.; Otteneder, M.B.; Singer, T.; Parrott, N. Characterization of Pharmacokinetics in the Göttingen Minipig with Reference Human Drugs: An In Vitro and In Vivo Approach. Pharm. Res. 2016, 33, 2565–2579.
- Shang, H.; Guo, K.; Liu, Y.; Yang, J.; Wei, H. Constitutive expression of CYP3A mRNA in Bama miniature pig tissues. Gene 2013, 524, 261–267.
- Skaanild, M.T.; Friis, C. Characterization of the P450 System in Göttingen Minipigs. Pharmacol. Toxicol. 1997, 80, 28–33.
- Souček, P.; Zuber, R.; Anzenbacherová, E.; Anzenbacher, P.; Guengerich, F.P. Minipig cytochrome P450 3A, 2A and 2C enzymes have similar properties to human analogs. BMC Pharmacol. 2001, 1, 11.
- Millecam, J.; De Clerck, L.; Govaert, E.; Devreese, M.; Gasthuys, E.; Schelstraete, W.; Deforce, D.; De Bock, L.; Van Bocxlaer, J.; Sys, S.; et al. The Ontogeny of Cytochrome P450 Enzyme Activity and Protein Abundance in Conventional Pigs in Support of Preclinical Pediatric Drug Research. Front. Pharmacol. 2018, 9, 470.
- Cazeneuve, C.; Pons, G.; Rey, E.; Treluyer, J.M.; Cresteil, T.; Thiroux, G.; D’Athis, P.; Olive, G. Biotransformation of caffeine in human liver microsomes from foetuses, neonates, infants and adults. Br. J. Clin. Pharmacol. 1994, 37, 405–412.
- Lacroix, D.; Sonnier, M.; Moncion, A.; Cheron, G.; Cresteil, T. Expression of CYP3A in the Human Liver-Evidence that the Shift between CYP3A7 and CYP3A4 Occurs Immediately After Birth. JBIC J. Biol. Inorg. Chem. 1997, 247, 625–634.
- Sonnier, M.; Cresteil, T. Delayed ontogenesis of CYP1A2 in the human liver. JBIC J. Biol. Inorg. Chem. 1998, 251, 893–898.
- Treluyer, J.M.; Gueret, G.; Cheron, G.; Sonnier, M.; Cresteil, T. Developmental expression of CYP2C and CYP2C-dependent activities in the human liver: In-vivo/in-vitro correlation and inducibility. Pharmacogenetics 1997, 7, 441–452.
- Treluyer, J.-M.; Jacqz-Aigrain, E.; Alvarez, F.; Cresteil, T. Expression of CYP2D6 in developing human liver. JBIC J. Biol. Inorg. Chem. 1991, 202, 583–588.
- Hu, S.X. Age-related change of hepatic uridine diphosphate glucuronosyltransferase and sulfotransferase activities in male chickens and pigs. J. Veter. Pharmacol. Ther. 2016, 40, 270–278.
- Millecam, J.; De Baere, S.; Croubels, S.; Devreese, M. In Vivo Metabolism of Ibuprofen in Growing Conventional Pigs: A Pharmacokinetic Approach. Front. Pharmacol. 2019, 10.
- Upreti, V.V.; Wahlstrom, J.L. Meta-analysis of hepatic cytochrome P450 ontogeny to underwrite the prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling. J. Clin. Pharmacol. 2015, 56, 266–283.
- Lu, H.; Rosenbaum, S. Developmental Pharmacokinetics in Pediatric Populations. J. Pediatr. Pharmacol. Ther. 2014, 19, 262–276.
- Brouwer, K.L.R.; Aleksunes, L.M.; Brandys, B.; Giacoia, G.P.; Knipp, G.T.; Lukacova, V.; Meibohm, B.; Nigam, S.K.; Rieder, M.; De Wildt, S.N.; et al. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin. Pharmacol. Ther. 2015, 98, 266–287.
- Schneckener, S.; Preuss, T.G.; Kuepfer, L.; Witt, J. A workflow to build PBTK models for novel species. Arch. Toxicol. 2020, 94, 3847–3860.
- Suenderhauf, C.; Tuffin, G.; Lorentsen, H.; Grimm, H.-P.; Flament, C.; Parrott, N. Pharmacokinetics of Paracetamol in Göttingen Minipigs: In Vivo Studies and Modeling to Elucidate Physiological Determinants of Absorption. Pharm. Res. 2014, 31, 2696–2707.
- Kesisoglou, F.; Balakrishnan, A.; Manser, K. Utility of PBPK Absorption Modeling to Guide Modified Release Formulation Development of Gaboxadol, a Highly Soluble Compound With Region-Dependent Absorption. J. Pharm. Sci. 2016, 105, 722–728.
- Shida, S.; Yamazaki, H. Human plasma concentrations of five cytochrome P450 probes extrapolated from pharmacokinetics in dogs and minipigs using physiologically based pharmacokinetic modeling. Xenobiotica 2015, 46, 759–764.
- Poulin, P.; Collet, S.H.; Atrux-Tallau, N.; Linget, J.-M.; Hennequin, L.; Wilson, C.E. Application of the Tissue Composition–Based Model to Minipig for Predicting the Volume of Distribution at Steady State and Dermis-to-Plasma Partition Coefficients of Drugs Used in the Physiologically Based Pharmacokinetics Model in Dermatology. J. Pharm. Sci. 2019, 108, 603–619.