The tumor microenvironment (TME) has become the focus of interest in cancer research and treatment. It includes the extracellular matrix (ECM) and ECM-modifying enzymes that are secreted by cancer and neighboring cells. The ECM serves both to anchor the tumor cells embedded in it and as a means of communication between the various cellular and non-cellular components of the TME. The cells of the TME modify their surrounding cancer-characteristic ECM. This in turn provides feedback to them via cellular receptors, thereby regulating, together with cytokines and exosomes, differentiation processes as well as tumor progression and spread. Matrix remodeling is accomplished by altering the repertoire of ECM components and by biophysical changes in stiffness and tension caused by ECM-crosslinking and ECM-degrading enzymes, in particular matrix metalloproteinases (MMPs). These can degrade ECM barriers or, by partial proteolysis, release soluble ECM fragments called matrikines, which influence cells inside and outside the TME.
- tumor microenvironment
- extracellular matrix
- inte
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
The tumor microenvironment (TME) describes the conditions within and in the vicinity of a solid tumor mass. It is shaped in an orchestrated manner by the oncogenically transformed cells and their neighboring tissue cells. It comprises cellular and noncellular constituents of macromolecular size and smaller molecules, as well as several biophysical parameters, such as pH, redox status [1], and mechanical tension within the tissue [2,3][2][3] (Figure 1). Among the small molecules, aberrant concentrations of redox potential-determining compounds, such as glutathione and reactive oxygen species (ROS) as well as extracellular ATP, characterize the TME [1].
Fibrillar and non-fibrillar proteins and rather amorphous proteoglycans together form the insoluble scaffold of the extracellular matrix [4,5][4][5] (Figure 1). The TME is also unique in its composition of soluble extracellular proteins, such as cytokines [6,7][6][7] and enzymes. Among the latter, extracellular matrix (ECM)-degrading matrix metalloproteinases (MMPs) and ECM-crosslinking lysyl-oxidases and transglutaminases characteristically contribute to the plasticity and distinction of the TME [8–11][8][9][10][11]. They are actively produced and remodel the ECM of the TME in a way, which influences cancer cells and their neighboring cells in a tumor-supportive manner. This review will shed light on the ECM of the TME and will take into account its TME-characteristic remodeling with a special emphasis on the MMPs. Moreover, it will summarize the current knowledge on the interactions of TME-embedded cells, both cancer and resident cells, with the ECM and the mutual effects on each other in maintaining tumor-supportive surroundings and in fostering metastasis.
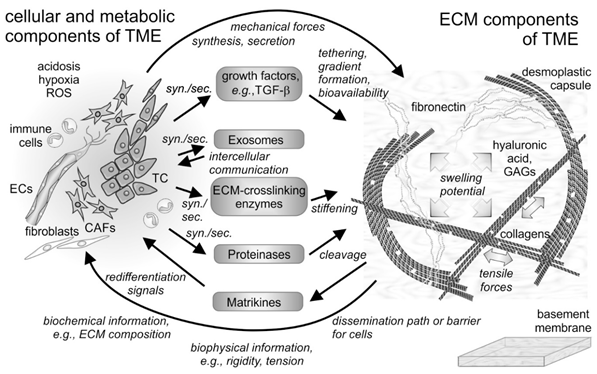
Figure 1. Cellular and non-cellular components of the tumor microenvironment (TME) and their interplay. In addition to the tumor cells (TCs), fibroblasts and their derivatives, the cancer-associated fibroblasts (CAFs), as well as ingrowing endothelial cells (ECs) and infiltrating immune cells are the cellular components of the tumor microenvironment (TME). The cells synthesize and secrete (syn./sec.) not only the extracellular matrix (ECM) components, but also growth factors, exosomes, and ECM-modifying enzymes, such as proteinases. Both cells and their secretion products interact with the fibrillar and non-fibrillar components of the ECM. Hyaluronic acid and glycosaminoglycan (GAG)-chains of proteoglycans increase the swelling potential of the interstitial space, which is counterbalanced by the tensile force-bearing fibrils of collagens, elastin and fibronectin. Modified by tethered growth factors, by crosslinking and cleaving enzymes, and by contractile forces, the ECM and its fragments with cytokine-like functions (matrikines) influence the cells within the TME in various ways. ROS, reactive oxygen species.
2. The Extracellular Matrix as a Key Component of the TME
The palpation of, e.g., the mammary gland [3], is a simple procedure to detect volume-demanding and stiffer tumor tissue [2,12][12]. Biophysical differences between normal tissue and tumor mass are caused by cell growth and the increased deposition of ECM components, known as desmoplasia, which is typically observed in healing wounds and fibrosis [13][13].
Most of the mass of solid tumors consists of ECM [14]. Having developed from collagen-rich stromal tissue, the TME is rich in collagens, especially if the tumor mass induces a desmoplastic reaction [13,15][15]. Collagens as the most abundant proteins of the human body crucially contribute to the scaffolding function of the ECM. The almost 30 members of the collagen family share several characteristics: (i) Their three chains consist of the repetitive Gly–X–Y amino acid sequence with X and Y being different amino acids, most frequently proline and hydroxyproline; (ii) They form a characteristic, staggered triple helix with the glycine residues of all triplet sequences in its center; (iii) They self-assemble into supramolecular structures, in which several triple-helical collagen molecules associate forming fibrils, networks, and other highly ordered aggregates [5,14,16][16]. Fibrils of type I collagen, together with collagen types III and V, bear the tensile forces within normal stromal tissue and in the TME of the tumor mass. They are preferentially deposited in desmoplastic environments, where the resident stromal cells are induced by tumor cells to produce and deposit collagen type I to form the stiff TME or a capsule surrounding the tumor mass [13,15][15]. Another collagen isoform, type IV collagen, along with collagens XV and XVIII, forms a network-like suprastructure, which is typical of basement membranes (BMs), the specialized sheet-like ECM that separates stromal tissue from other tissues. As it confines cells to their respective tissue type, its breaching by malignant cells is a hallmark of cancer [17].
Spanning the interstitial stroma, collagen fibrils provide an ideal path for cell migration and promote cancer cell dissemination along these fibrils (Figure 2). In contrast, the meshwork of stromal collagen fibrils and the desmoplastic capsule around the tumor mass like the type IV collagen network of basement membranes are extremely dense and impede tumor cell infiltration [18,19][18][19]. These ECM barriers are overcome by the cancer cells or their accompanying CAFs by the cleavage of collagen with particular collagenases [20].
In addition to the network-forming collagens, laminins, which form a family of about 20 members, are typical constituents of basement membranes [21–23][21][22][23]. Their N termini and the C terminus formed by the globular G domain of the laminin α chain protrude from an α-helical coiled coil [21]. Although laminins are normally exclusively found in BMs, some types of laminins, such as laminin-332, also occur ectopically within the TME [24], but their role in the TME has not yet been fully deciphered.
Collagen fibrils are often found together with elastin and fibulin containing elastic fibrils [25], which, due to their reversible elasticity, allow the resilience of the ECM. Interestingly, most of the body’s elastin is formed pre- and early postnatally but hardly in the adult body [26]. However, some cancer entities stimulate and reinitiate elastin production and deposition, which is known as elastosis [25]. Similarly noteworthy, elastin degradation peptides (EDPs) are released in the TME, which stimulate tumor cell growth and progression via different receptors [27] (Figure 2).
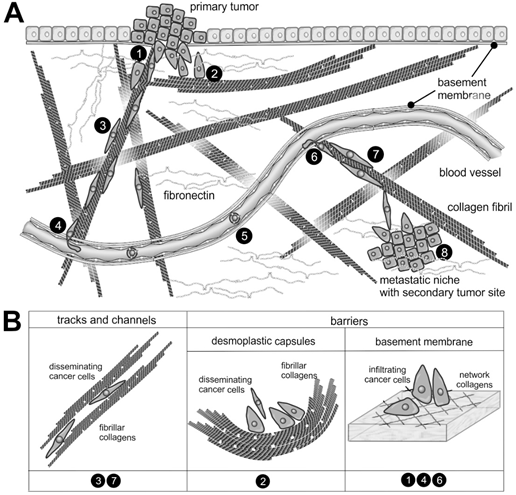
Figure 2. The supramolecular structure of collagen as a substrate or impediment of cancer cell dissemination: (A) The metastatic cascade of malignant carcinoma cells includes the penetration of the epithelial basement membrane (no.1); Solid tumors within the stroma tissue are often surrounded by a desmoplastic collagen capsule, which impedes cancer cell migration (no.2); Through the interstitial stroma, cancer cells utilize collagen-rich fibrils to quickly reach a nearby blood vessel (no.3); There, they intravasate through the endothelial basement membrane (no.4); When blood-borne, they float with the blood stream, mostly sheltered by platelets (no.5); To extravasate, they attach to the vessel wall of a distant organ and again breach the endothelial basement membrane (no.6); Moving quickly along collagen-rich fibrils (no.7), they reach the metastatic niche (no.8), where they grow into a metastasis. (B) The different dissemination-supporting and impeding functions of the collagen suprastructures are highlighted for the different steps of the metastatic cascade. The cancer cells experience, particularly by integrins, the ECM as dissemination-supporting tracks and channels, especially if migration and fibrils point into the same direction and if the fibril network is not too dense. In contrast, the dense array of collagen fibrils in desmoplastic capsules and of network-forming collagens within the basement membrane impedes cancer cell progression and requires the use matrix-metalloproteinases. Moreover, the orientation of collagen fibrils within the desmoplastic capsule is mostly perpendicular to the direction of dissemination [28] [28].
Fibronectin is another scaffold-forming glycoprotein found in BMs as well as in the ECM of the TME [29[29][30],30], with distinct splice variants being produced in the TME [31]. Fibronectin consists of two disulfide-linked protein chains with a characteristic modular character of fibronectin repeats of types I, II, and III with about 30, 60, and 90 amino acids, respectively. The type I and II repeats allow the formation of disulfide-crosslinked supramolecular fibronectin networks in the tissue stroma [30]. Fibronectin isoforms with the extra domains ED-A and ED-B are normally expressed under the control of mainly TGF-β during embryonic development and wound repair but also in the hypoxic TME [32,33][32][33]. Thus, ED-A- and/or ED-B- fibronectin are employed as a marker to image tumor nodes [34]. Other glycoproteins marking a tumor-modified stroma are tenascins-C and W [35–37][35][36][37]. Like the other two members of the tenascin family [38], they are disulfide-crosslinked homotrimers, each consisting of three chains forming an α-helical coiled coil. With their ECM-typical EGF-like and type III fibronectin domains, and a C-terminal fibrinogen globe module, they perform various functions in wound healing and tumor progression [36]. These tenascin isoforms or fragments thereof can also be released from the primary tumor into the blood circulation and precondition distant sites as premetastatic niches [39]. Thus, the blood levels of tenascins are a diagnostic tumor marker [40]. Their abundance in the TME is tested for diagnostic imaging and therapeutic exploitation [41]. Tenascins and other pericellular ECM proteins, such as periostin [42], galectins [43], small integrin-binding ligand N-linked glycoproteins (SIBLINGs), secreted protein acidic and rich in cysteine (SPARC), thrombospondin, angiopoietin-like proteins, certain proteoglycans, and CCN family members are referred to as matricellular proteins [44]. Rather than performing scaffolding functions, they modulate the supramolecular architecture of the collagen and fibronectin network [42] and they regulate cellular behavior within the TME [37]. For example, CCNs regulate the proliferation and migration of ECM scaffold-embedded cells [44,45][44][45].
Functionally similarly versatile are glycosaminoglycans (GAGs), and the protein- and sulfate-free hyaluronic acid (HA) made up of N-acetylglucosamine and glucuronic acid [46[46][47],47], as well as sulfated GAG-chain containing proteoglycans, such as heparan sulfate and keratan sulfate, and galactosamine-containing chondroitin sulfate and dermatan sulfate [4,5,48,49][48][49]. According to their location, proteoglycans are divided into extracellular, membrane-bound, and intracellular proteoglycans. The extracellular group includes the hyaluronic acid-binding hyalectans (e.g., aggrecan, versican with four different splice variants, neurocan, and brevican), the small leucine-rich proteoglycans (SLRPs, e.g., decorin, biglycan, fibromodulin, and lumican), and the BM-located pericellular proteoglycans (e.g., perlecan, agrin, and collagen XVIII). Perlecan, agrin, and the network-forming type VIII collagen bear heparan sulfate GAG chains [48], while the hyalectans and SLRPs are decorated with different numbers of covalently attached chondroitin sulfate, sometimes in combination with additional dermatan sulfate chains (decorin) or keratan sulfate chains (aggrecan, fibromodulin, lumican) [49]. The extracellular proteoglycans non-covalently link up with the scaffold of fibril-forming ECM components in different ways. Type IX collagen decorates the surface of type II collagen fibrils, while type XVIII collagen forms the BM-typical chicken wire network together with type IV collagen. Although alloyed into collagen fibrils, they are also proteoglycans due to their chondroitin sulfate and heparan sulfate GAG-chains, respectively [4,5]. Alternatively, several proteoglycans, such as decorin and other SLRPs, interact via their protein cores with type I collagen-containing fibrils and thus control the suprastructure of the ECM scaffold [46]. Moreover, specific type III repeats of fibronectin, certain G-domains of laminin α-chains, tenascins and thrombospondins harbor heparan sulfate binding sites, thereby allowing protein-carbohydrate binding interactions to connect the “amorphous” proteoglycans with higher suprastructure-forming ECM components [5]. Likewise, the hyalectans have a carbohydrate-binding domain, which allows their binding to hyaluronic acid.
Proteoglycans with their GAG chains also specifically tether growth factors and assist in presenting them to the respective cellular receptors. Such tethering of growth factors and cytokines stabilizes the formation of spatial gradients, which are indispensable for normal development and also contribute to pathologic processes. For example, in tumor angiogenesis, endothelial cells (ECs) follow a VEGF gradient that is stabilized by tethering to heparan sulfate GAG chains [47,48][47][48].
References
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448, doi:10.1016/j.ajps.2019.08.003.
- Emon, B.; Bauer, J.; Jain, Y.; Jung, B.; Saif, T. Biophysics of Tumor Microenvironment and Cancer Metastasis—A Mini Review. Comput. Struct. Biotechnol. J. 2018, 16, 279–287, doi:10.1016/j.csbj.2018.07.003.
- Mohammadi, H.; Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 2018, 20, 766–774, doi:10.1038/s41556-018-0131-2.
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55, doi:10.1016/j.matbio.2015.02.003.
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27, doi:10.1016/j.addr.2015.11.001.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204, doi:10.1186/s13046-020-01709-5.
- Zhao, H.; Wei, J.; Sun, J. Roles of TGF-beta signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020, 89, 107101, doi:10.1016/j.intimp.2020.107101.
- Chitty, J.L.; Setargew, Y.F.I.; Cox, T.R. Targeting the lysyl oxidases in tumour desmoplasia. Biochem. Soc. Trans. 2019, 47, 1661–1678, doi:10.1042/bst20190098.
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83, doi:10.1016/j.critrevonc.2019.02.010.
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729, doi:10.3390/cancers11050729.
- Lentini, A.; Abbruzzese, A.; Provenzano, B.; Tabolacci, C.; Beninati, S. Transglutaminases: Key regulators of cancer metastasis. Amino Acids 2013, 44, 25–32, doi:10.1007/s00726-012-1229-7.
- Leight, J.L.; Drain, A.P.; Weaver, V.M. Extracellular Matrix Remodeling and Stiffening Modulate Tumor Phenotype and Treatment Response. Ann. Rev. Cancer Biol. 2017, 1, 313–334, doi:10.1146/annurev-cancerbio-050216-034431.
- Martins Cavaco, A.C.; Damaso, S.; Casimiro, S.; Costa, L. Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev. 2020, 39, 603–623, doi:10.1007/s10555-020-09888-5.
- Bourgot, I.; Primac, I.; Louis, T.; Noel, A.; Maquoi, E. Reciprocal Interplay Between Fibrillar Collagens and Collagen-Binding Integrins: Implications in Cancer Progression and Metastasis. Front. Oncol. 2020, 10, 1488, doi:10.3389/fonc.2020.01488.
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309, doi:10.1186/s12967-019-2058-1.
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978, doi:10.1101/cshperspect.a004978.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674, doi:10.1016/j.cell.2011.02.013.
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011, 21, 736–744, doi:10.1016/j.tcb.2011.09.006.
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084, doi:10.1083/jcb.201210152.
- Overall, C.M. Matrix metalloproteinase substrate binding domains, modules and exosites. Overview and experimental strategies. Methods Mol. Biol. 2001, 151, 79–120.
- Aumailley, M. The laminin family. Cell Adh. Migr. 2013, 7, 48–55, doi:10.4161/cam.22826.
- Halfter, W.; Oertle, P.; Monnier, C.A.; Camenzind, L.; Reyes-Lua, M.; Hu, H.; Candiello, J.; Labilloy, A.; Balasubramani, M.; Henrich, P.B.; et al. New concepts in basement membrane biology. FEBS J. 2015, 282, 4466–4479, doi:10.1111/febs.13495.
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adh. Migr. 2013, 7, 56–63, doi:10.4161/cam.21831.
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The Interaction between Laminin-332 and alpha3beta1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers 2018, 11, 14, doi:10.3390/cancers11010014.
- Wang, Y.; Song, E.C.; Resnick, M.B. Elastin in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 1–16, doi:10.1007/978-3-030-48457-6_1.
- Muiznieks, L.D.; Weiss, A.S.; Keeley, F.W. Structural disorder and dynamics of elastin. Biochem. Cell. Biol. 2010, 88, 239–250, doi:10.1139/o09-161.
- Heinz, A. Elastases and elastokines: Elastin degradation and its significance in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 252–273, doi:10.1080/10409238.2020.1768208.
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198, doi:10.1007/s10585-019-09966-1.
- White, E.S.; Muro, A.F. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011, 63, 538–546, doi:10.1002/iub.493.
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27, doi:10.3390/cells9010027.
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Front. Oncol. 2020, 10, 641, doi:10.3389/fonc.2020.00641.
- Lee, S.H.; Lee, Y.J.; Han, H.J. Role of hypoxia-induced fibronectin-integrin beta1 expression in embryonic stem cell proliferation and migration: Involvement of PI3K/Akt and FAK. J. Cell. Physiol. 2011, 226, 484–493, doi:10.1002/jcp.22358.
- Ryu, M.H.; Park, H.M.; Chung, J.; Lee, C.H.; Park, H.R. Hypoxia-inducible factor-1alpha mediates oral squamous cell carcinoma invasion via upregulation of alpha5 integrin and fibronectin. Biochem. Biophys. Res. Commun. 2010, 393, 11–15, doi:10.1016/j.bbrc.2010.01.060.
- Sollini, M.; Boni, R.; Traino, A.C.; Lazzeri, E.; Pasqualetti, F.; Modeo, L.; Mariani, G.; Petrini, M.; Erba, P.A. New approaches for imaging and therapy of solid cancer. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 168–183.
- Degen, M.; Brellier, F.; Kain, R.; Ruiz, C.; Terracciano, L.; Orend, G.; Chiquet-Ehrismann, R. Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res. 2007, 67, 9169–9179, doi:10.1158/0008-5472.CAN-07-0666.
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310, doi:10.1007/s12079-009-0075-1.
- Scherberich, A.; Tucker, R.P.; Degen, M.; Brown-Luedi, M.; Andres, A.C.; Chiquet-Ehrismann, R. Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene 2005, 24, 1525–1532, doi:10.1038/sj.onc.1208342.
- Brellier, F.; Tucker, R.P.; Chiquet-Ehrismann, R. Tenascins and their implications in diseases and tissue mechanics. Scand. J. Med. Sci. Sports 2009, 19, 511–519, doi:10.1111/j.1600-0838.2009.00916.x.
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874, doi:10.1038/nm.2379.
- Degen, M.; Brellier, F.; Schenk, S.; Driscoll, R.; Zaman, K.; Stupp, R.; Tornillo, L.; Terracciano, L.; Chiquet-Ehrismann, R.; Ruegg, C.; et al. Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int. J. Cancer 2008, 122, 2454–2461, doi:10.1002/ijc.23417.
- Spenle, C.; Saupe, F.; Midwood, K.; Burckel, H.; Noel, G.; Orend, G. Tenascin-C: Exploitation and collateral damage in cancer management. Cell Adh. Migr. 2015, 9, 141–153, doi:10.1080/19336918.2014.1000074.
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039, doi:10.1074/jbc.M109.051961.
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, doi:10.1084/jem.20182041.
- Thakur, R.; Mishra, D.P. Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74, doi:10.1016/j.pharmthera.2016.09.002.
- Leask, A. Conjunction junction, what’s the function? CCN proteins as targets in fibrosis and cancers. Am. J. Physiol. Cell Physiol. 2020, 318, C1046–C1054, doi:10.1152/ajpcell.00028.2020.
- Chen, S.; Birk, D.E. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J. 2013, 280, 2120–2137, doi:10.1111/febs.12136.
- Xie, M.; Li, J.P. Heparan sulfate proteoglycan—A common receptor for diverse cytokines. Cell. Signal. 2019, 54, 115–121, doi:10.1016/j.cellsig.2018.11.022.
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycan Signaling in Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 6588, doi:10.3390/ijms21186588.
- Mellai, M.; Casalone, C.; Corona, C.; Crociara, P.; Favole, A.; Cassoni, P.; Schiffer, D.; Boldorini, R. Chondroitin Sulphate Proteoglycans in the Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 73–92, doi:10.1007/978-3-030-48457-6_5.