G protein-coupled receptor (GPCR) kinases (GRKs) play an important role in the regulation of signaling of GPCRs that bind neurotransmitters. The canonical model of GPCR desensitization posits that GRKs phosphorylate ligand-activated GPCRs, and this phosphorylation prepares receptors for the high-affinity binding of arrestin proteins. Arrestin binding prevents further G protein coupling, promotes receptor internalization, and initiates and/or facilitates specific branches of signaling. Existing data suggest that the role of GPCR phosphorylation by GRKs is distinct in different receptors. The relationship between G protein- and arrestin-mediated signaling on the one hand, and therapeutic and side effects of drugs on the other, is more complex than is widely believed. Also, the relationship between rapid (minutes to hours) GRK/arrestin-mediated regulation and long-term (days to weeks) neural plasticity remains to be elucidated.
- GRK
- GPCR
- neurotransmitter
- arrestin
1. Introduction: GRKs in GPCR Signaling and Trafficking
Activation-dependent phosphorylation of rhodopsin was discovered in the early 1970s [1][2][1,2], long before it became clear that rhodopsin belongs to the family of G protein-coupled receptors (GPCRs). Comparison of the primary structure and membrane organization of rhodopsin [3] and β2-adrenergic receptor (β2AR) [4] [4] demonstrated that both belong to the same protein family, now known as the GPCR superfamily, of which humans express ~800 different subtypes. These receptors are also called 7TMRs, as all GPCRs contain seven transmembrane α-helices. Rhodopsin kinase (modern systematic name GRK1, which stands for G protein-coupled receptor kinase 1 [5]) was the first GPCR kinase (GRK) discovered. It was shown to selectively bind and phosphorylate light-activated rhodopsin [6][7][6,7]. Later, the Kuhn group demonstrated that rhodopsin phosphorylation is necessary to quench its signaling [8]. In the same year, the Lefkowitz group reported that β2AR is phosphorylated by a cAMP-independent kinase and that this phosphorylation facilitates receptor desensitization [9]. As this kinase was discovered via its ability to phosphorylate β2AR, when cloned, it was called β2-adrenergic receptor kinase, or βARK for short (modern systematic name GRK2) [10]. GRK2 was also shown to phosphorylate rhodopsin in a strictly activation-dependent manner, exactly like rhodopsin kinase [11], suggesting that both GRKs are specific for active GPCRs. When rhodopsin kinase was cloned [12], it was found to have a structure similar to that of the kinase that phosphorylates agonist-activated β2AR [10]. The mechanism underlying the specificity of GRKs for active GPCRs was subsequently established: the activation of GRK1 required physical interaction with the active rhodopsin [13]. The same activation mechanism was demonstrated for non-visual GRKs with β2AR [14] [14] and D2R [15].
Rhodopsin phosphorylation was shown to increase the binding of a 48-kDa protein [16] [16] (later termed visual or rod arrestin; systematic name arrestin-1). The binding of this protein was shown to be necessary to stop rhodopsin coupling to its cognate G protein, transducin [8]. It turned out that highly purified β2AR kinase has a limited effect on β2AR coupling to its cognate G protein, Gs, suggesting that a non-visual arrestin homologue is necessary for desensitization [17]. Both arrestin-1 [18] [18] and its non-visual homologue [19] were subsequently cloned. These proteins are highly homologous. However, arrestin-1 and its homologue demonstrated clear preference for rhodopsin and β2AR, respectively [20]. Therefore, the non-visual protein was originally termed β-arrestin (systematic name arrestin-2).
In genetically modified mice, it was shown that the absence of either GRK1 [21] [21] or arrestin-1 [22] results in abnormally prolonged rhodopsin signaling. These and other findings resulted in the general model of two-step homologous (specific for the receptor that was activated) GPCR desensitization: the active receptor is phosphorylated by a GRK (seven GRK subtypes are expressed in most vertebrates [5][23][5,23]), whereupon arrestin (four subtypes are expressed in most vertebrates [24]) binds to it and stops the signaling by direct competition with the G protein [25][26] [25,26] (reviewed in [27]) (Figure 1). Non-visual arrestins bound to phosphorylated GPCRs were shown to directly interact with the key components of the internalization machinery of the coated pit, clathrin [28], and clathrin adaptor AP2 [29], so that GPCR phosphorylation and subsequent arrestin binding promote receptor endocytosis. While the role of both GPCR phosphorylation and arrestin binding actually differs for various receptor subtypes (Figure 2), the simplified general paradigm posits that GRKs play a critical role in the two processes that reduce cell responsiveness: precluding G protein coupling and facilitating receptor removal from the plasma membrane. These are the key GRK effects on the signaling of most GPCRs, including the neurotransmitter receptors belonging to the GPCR family that are discussed below. The amount of information available regarding the effects of GRK phosphorylation, particularly in biologically relevant in vivo models, varies significantly for different receptors. While several independent studies have been performed with opioid receptors, including the use of knockin mice expressing receptor mutants where some or all potential GRK phosphorylation sites were eliminated, other neurotransmitter GPCRs have been studied much less comprehensively. As this review is based on experimental evidence, opioid receptors will be discussed in greater detail than other GPCRs that bind neurotransmitters.
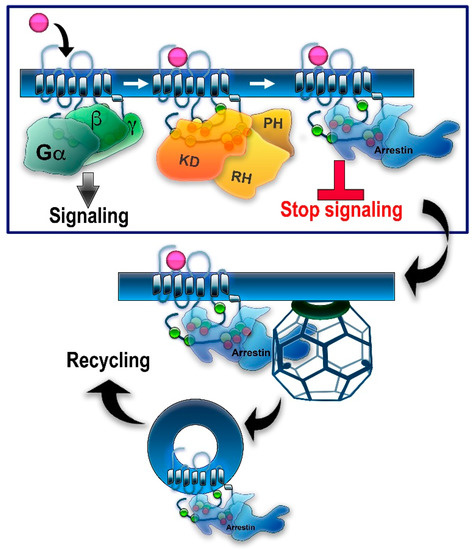
Figure 1.
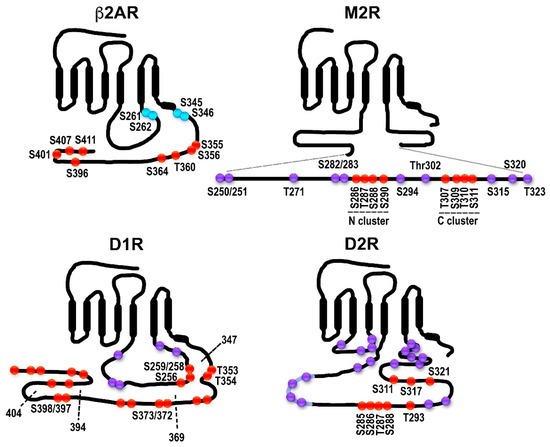
Figure 2. Distribution of potential and established GRK phosphorylation sites in selected GPCRs. The classical model does not necessarily apply to all GPCRs: in some cases, receptor phosphorylation is not necessary or plays a minor role in arrestin binding; some arrestin-associated receptors do not internalize via coated pits; for others, arrestins appear to mediate receptor internalization, but not desensitization, etc. β2-adrenergic receptor (β2AR). The sites in the human β2AR are shown. The sites of PKA phosphorylation are shown in blue. The sites of GRK phosphorylation are shown in red. All GRK targets are localized in the C-terminus, as in rhodopsin, β1AR, opioid and cannabinoid receptors, and many other GPCRs. Muscarinic M2 receptor (M2R). The sequence and GRK phosphorylation sites in the human M2R are shown. All putative sites are located in the 3rd cytoplasmic loop; the actual phosphorylation sites have been localized to the central part of the 3rd loop (Ser250-Thr323 shown here as an insert). The sites in the two characterized clusters are shown in red, the other sites in magenta. Two clusters of phosphorylatable residues (red) with different functions were described. Thr307-Ser311 (C cluster) appears to be necessary for desensitization; when the N cluster (residues Ser286-Ser290) is mutated to alanines, the receptor still desensitizes. Arrestin binding depends on the C-cluster [30]. Internalization is promoted by phosphorylation of either cluster [30][31][32]. Other potential phosphorylation sites within the region are shown in purple. Dopamine D1 receptor (D1R). D1R has sites in both the C-terminus and the 3rd cytoplasmic loop. The rat D1R is shown. The sites labeled in red have been shown to be phosphorylated in an agonist-dependent manner either in truncation experiments or via mutations to alanines [33]. Truncations used in [33] are shown as dotted lines with the last residue remaining labeled. Other potential phosphorylation sites are shown in purple. Dopamine D2 receptor (D2R). The rat D2R is shown. All phosphorylatable sites are in the 3rd cytoplasmic loop. Eight sites phosphorylated in an agonist-dependent manner by GRKs are shown in red [34]. Some of the other potential phosphorylation sites are shown in purple.
2. From Neurotransmitter Receptor Regulation to Neural Adaptation: The Role of GRKs
It is not difficult to imagine that altered availability and/or function of GRKs, leading to impaired receptor desensitization/trafficking, would result in enhanced or reduced responsiveness to agonists, as in cultured cells. However, chronic treatment with neurotropic drugs often results in long-term neural plasticity, altering the responsiveness to these drugs, which develops over hours or days, in contrast to the much faster timescale of receptor phosphorylation and arrestin recruitment, both of which occur within minutes in cultured cells. This long-lasting plasticity is initiated by agonist stimulation of the neurotransmitter receptors, which would initially engage GRKs, and it is a legitimate—and intriguing—question whether GRK-dependent phosphorylation of the neurotransmitter receptors plays a role in the development of long-term neural plasticity.
2.1. GRKs in the Regulation of Acute Responsiveness to Neural Stimulation
Loss of GRK5 results in supersensitivity to muscarinic M2 receptor stimulation [35][36]. GRK5 deficiency impairs desensitization of M2/M4 autoreceptors, causing inhibition of the hippocampal acetylcholine release and cholinergic hypofunction [37]. The acetylcholine deficiency caused by the loss of one copy of the GRK5 gene in mice overexpressing β-amyloid precursor protein (APP) with the Swedish mutation (Tg2576) exacerbates the accumulation of β-amyloid [38]. Selective deletion of GRK2 in the brain cholinergic neurons caused reduced sensitivity to the effects of the muscarinic agonist oxotremorine such as hypothermia, hypolocomotion, salivation, and antinociception [39]. The hypothermia is mediated by M2 receptors, hypolocomotion is caused by the action via M1 and M4 receptors, and salivation is governed by M1, M3, and M4 receptors [40][41][42][43]. Since the GRK2 loss occurred in cholinergic neurons, it is likely to primarily affect muscarinic autoreceptors such as M2/M4. Unfortunately, the location within the brain circuitry remains unknown, which makes it impossible to interpret these data at the molecular level, since there are many cholinergic cell groups with diverse functions. Collectively, these studies have highlighted an important role of GRKs in the regulation of receptor responsiveness to stimulation. The functional role of GRK-mediated phosphorylation of specific receptors at precisely defined sites within the brain circuitry has not yet been addressed.
The exact relation between GRK-mediated receptor phosphorylation and in vivo adaptations to receptor stimulation has been studied in some detail in the case of opioid receptors. Physiologically, opioids induce analgesia in humans and animals, and classic opioid drugs exert their effects via MOR. If opioid analgesia is mediated by G protein activation via MOR, desensitization of the receptor would serve to limit the extent and duration of the analgesic response. Indeed, knockin mice expressing MOR with Ser 375 mutated to alanine (Ser375Ala), which cannot be phosphorylated, showed greater antinociceptive response to morphine and fentanyl [44]. In knockin mice with multiple serines and threonines in MOR mutated to alanines, which made the receptor increasingly unable to be phosphorylated, recruit arrestins, and be desensitized, antinociceptive responses to morphine and fentanyl were significantly enhanced and prolonged [45]. Mice lacking GRK3 showed unchanged antinociceptive responses [46][47], presumably because GRK2 was still available to phosphorylate MOR. Unfortunately, the role of GRK2 has not so far been examined using this approach, because global GRK2 knockout in mice is embryonically lethal. Interestingly, the loss of GRK5 had the opposite effect: GRK5 knockout mice demonstrated diminished antinociception as compared to wild type [46]. This latter result is counterintuitive, suggesting that some action(s) of GRK5 is involved other than its role in receptor phosphorylation/desensitization. Similar to the opioid receptors, impaired desensitization of cannabinoid receptors results in enhanced acute responsiveness to stimulation. In mice with two prime phosphorylation sites (Ser426 and Ser430) in the C-terminus of the CB1 receptor mutated to alanines, acute sensitivity to Δ9-THC was increased. Desensitization and downregulation of CB1 in the spinal cord was absent. In autaptic cultured hippocampal neurons, endocannabinoid responses were enhanced and their desensitization reduced [48].
2.2. GRK-Mediated Rapid Desensitization in Long-Term Neural Adaptations
Generally speaking, chronic tolerance appears to involve GRK-dependent phosphorylation of opioid receptors enabling arrestin recruitment, and when these functions are compromised, tolerance is diminished. The mechanism of this effect is intriguing, since the timings of these events—GRK/arrestin-dependent homologous desensitization and tolerance—are so different (discussed here [49]). However, the data do indicate the requirement for GRK-dependent receptor phosphorylation in the development of tolerance. Speaking specifically about MOR, tolerance to high-efficacy MOR agonists seems to depend on MOR phosphorylation at Ser375, which is the key residue for the initiation of the phosphorylation cascade leading to MOR desensitization/internalization. However, tolerance to morphine seems to involve additional mechanisms, although phosphorylation of multiple MOR residues appears to be a contributing factor.
In mice with the Ser426 and Ser430 phosphorylation sites in the C-terminus of the CB1 receptor mutated to alanines, dependence on Δ9-THC was increased, but tolerance delayed, which was accompanied by enhanced acute responses [48]. Tolerance to the antinociceptive effect of WIN55,212–2, another synthetic cannabinoid agonist, was also delayed in these mice [50]. Thus, in the case of cannabinoid receptors, tolerance appears to involve GRK phosphorylation and likely subsequent arrestin recruitment, as in opioid receptors. However, the time course of physiological tolerance to opioids and cannabinoids (days to weeks) is much longer than of receptor phosphorylation and subsequent arrestin binding (minutes) or even receptor internalization and downregulation (hours). Thus, it appears that receptor phosphorylation and/or subsequent formation of the receptor–arrestin complex initiates longer-term regulatory processes in neurons that are not apparent in cell culture models.
2.1. GRKs in the Regulation of Acute Responsiveness to Neural Stimulation
2.2. GRK-Mediated Rapid Desensitization in Long-Term Neural Adaptations
2.3. Neurotropic Drugs: To Bias or Not to Bias?
All therapeutically active drugs have side effects. Since the discovery of arrestin-mediated signaling by GPCRs, the idea of biased signaling, i.e., signaling engaging only one branch of the GPCR pathway, either G protein or arrestin, has gained popularity. For the biased signaling to be possible, the two branches, the G protein- and arrestin-mediated, should be independent, and originally they were believed to be. There are obvious limitations to this independence. The fact that GRKs 2 and 3 use two products of G protein activation, Gβγ [51][52][53] [60,61,62] and activated Gαq/11 [54][101], for membrane localization suggests that G protein activation likely plays a role in GPCR phosphorylation by these GRKs, which is often necessary for arrestin binding. This might limit what arrestins can do in the absence of G protein activation. On the other hand, GRKs 4/5/6 have C-terminal lipid modifications and/or specific sequences mediating their attachment to the membrane independently of G proteins (reviewed in [5]). Thus, phosphorylation of the same GPCRs by GRKs 5/6 instead of GRKs 2/3 might bypass the need for G protein and enable arrestin recruitment and arrestin-mediated signaling. Recently, a question arose as to whether G protein activity is required for arrestin-mediated signaling in some capacity other than for the GRK recruitment to the receptor. It has been reported that the presence of functional G proteins is indispensable for arrestin-mediated activation of the ERK pathway [55][165]. In cells, the ERK pathway is activated via G protein as well as arrestin-dependent mechanisms [56][57][166,167], but it can be activated exclusively via G proteins independently of arrestins [58][168]. Whether the reverse is true has not yet been unambiguously determined (see short discussion in [59][169]). The issue of the potential and limitations of G protein vs. arrestin bias of GPCR ligands was recently discussed in depth [60][148].
On the physiological side, the idea is based on the notion that the therapeutic action might be mediated by one signaling branch and the side effects by another. The attraction of this theory is obvious but the experimental basis is limited (discussed in [60][148]). Arguably, nowhere has this theory received more attention than in the field of opioid therapy. Opioid drugs remain the mainstay of pain management. Their utility is limited, however, not only by the tolerance that develops upon long-term use but also by multiple unwanted side effects including life-threatening respiratory depression, gastrointestinal disturbances, and addiction. Since GPCRs signal via both G protein- and arrestin-mediated pathways, it has been suggested that the therapeutic antinociceptive effects of opioid drugs are mediated by G proteins, whereas arrestin-dependent signaling is responsible for the side effects. This notion is based on studies of G protein-biased opioid agonists such as PZM21 [61] [170] and TRV-130 [62][171]. TRV130 induced less MOR phosphorylation and arrestin recruitment than unbiased agonists. It is a potent analgesic with less evident gastrointestinal dysfunction and respiratory suppression than morphine [62][63][171,172]. However, direct proof that the arrestin-mediated signaling that takes place following MOR phosphorylation and arrestin recruitment is responsible for the opioid side effects is lacking. It is important to note that in many cases—clearly in the case of opioid receptors, for which phosphorylation by GRKs is a prerequisite for arrestin recruitment—bias for or away from arrestin is in reality a bias for or away from specific GRKs. What hampers the meaningful discussion of biased signaling is the lack of information on the phosphorylation patterns of the most relevant receptors and their potential barcoding by GRK isoforms. A recent important study using knockin mice expressing phosphorylation-deficient MOR with all or nearly all phosphorylation sites eliminated demonstrated that the side effects of both morphine and fentanyl were, if anything, exacerbated [45][158]. This effect was accompanied by enhanced analgesia and diminished tolerance. Furthermore, the EC50 values for morphine analgesia and respiratory depression/constipation across lines with different phosphorylation levels are highly correlated (R2 > 0.9), suggesting that these effects are not independent. These experiments strongly suggest that all major physiological effects of MOR activation, both good and bad from our perspective, are largely mediated by G proteins.