Fanconi anemia (FA), a chromosomal instability syndrome, is caused by inherited pathogenic variants in any of 22 FANC genes, which cooperate in the FA/BRCA pathway. This pathway regulates the repair of DNA interstrand crosslinks (ICLs) through homologous recombination. In FA proper repair of ICLs is impaired and accumulation of toxic DNA double strand breaks occurs. To repair this type of DNA damage, FA cells activate alternative error-prone DNA repair pathways, which may lead to the formation of gross structural chromosome aberrations of which radial figures are the hallmark of FA, and their segregation during cell division are the origin of subsequent aberrations such as translocations, dicentrics and acentric fragments. The deficiency in DNA repair has pleiotropic consequences in the phenotype of patients with FA, including developmental alterations, bone marrow failure and an extreme risk to develop cancer.
- chromosomal instability
- FA pathway
- radial figure
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Fanconi anemia (FA) is a rare disease with an incidence of 1–5 per million of births and is the most commonly inherited bone marrow failure syndrome [1]. FA is caused by the failure of the Fanconi anemia/breast cancer (FA/BRCA) pathway [2]; thus far, 22 genes (FANCA to FANCW) that participate in this pathway have been identified. Germline pathogenic variants (PV) in any one of these genes are the origin of this disease [3]. PV show an autosomal recessive inheritance for 20 of these genes, autosomal dominant inheritance has been shown for one gene (FANCR/RAD51) and X-linked inheritance for another gene (FANCB). Inherited PV in the FANCA, FANCC or FANCG genes account for approximately 90% of FA cases, whereas the other 19 FANC genes account for the remaining 10% [3]. The FA/BRCA pathway is involved in the proper functioning of various cellular processes; one of its most important functions is during the repair of DNA interstrand crosslink (ICL), lesions that covalently join the two DNA strands and impair DNA replication and transcription. In everyday life, we are exposed to sources of ICL-inducing agents that can be both endogenous resulting from cellular metabolism or exogenous due to environmental, occupational or personal exposure habits. Some calculations suggest that in the steady state every cell could carry around 37,000 lesions per genome, and that ICLs account for approximately 1500 of these lesions, whose origin can be bi-functional chemicals related to formaldehyde, acetaldehyde, acrolein and in smaller proportions crotonaldehyde [4]. ICLs of exogenous origins are more difficult to assess since for all the agents with the capacity to induce them, only a small fraction (typically 1–5%) will be ICLs, while the majority of the induced DNA damage will be monoadducts or intrastrand crosslinks [5]. Most of this damage is successfully repaired via the FA/BRCA pathway.
Failure of the FA/BRCA pathway has consequences at various levels of complexity: (1) at the chromosomal level by the presence of numerical and structural chromosomal instability; (2) at the cellular level resulting in increased cell death, alteration of the cell cycle, high sensitivity to oxidative damage and to DNA cross-linking agents, both exogenous such as chemotherapeutic drugs, e.g. cis-platinum, mitomycin C or diepoxybutane, as well as endogenous aldehydes, a product of cell metabolic activities [6,7][6][7]; and (3) at the clinical level, where patients with FA present three main features: developmental abnormalities, bone marrow failure and an increased risk of cancer [8].
2. Double Strand Breaks Are at the Center of Chromosome Aberrations in FA
2.1. Involvement of FA/BRCA Pathway in DNA Repair
The protein products of the FANC genes collaborate in the FA/BRCA pathway to protect DNA replication fork and repair ICLs [1]. ICLs are dangerous lesions that prevent the opening of the double stranded DNA for transcription and replication. For ICLs repair, the cell needs to use several DNA repair converging mechanisms, and the FA/BRCA pathway is tasked with coordinating them to process the ICL into byproduct lesions, remarkably a DSB which is to be preferentially repaired in an error-free way. Individual functions of FANC proteins during this assemblage appear in Table 1.
Table 1. Fanconi anemia genes involved in FA/BRCA pathway 1.
|
FANC Gene/Alias |
Cytogenetic Location |
Function of the FANC Protein |
|
FANCA |
16q24.3 |
FA core complex |
|
FANCB |
Xp22.2 |
FA core complex |
|
FANCC |
9q22.32 |
FA core complex |
|
FANCD1/BRCA2 |
13q13.1 |
Homologous recombination. Enable RAD51 to displace RPA from ssDNA. |
|
FANCD2 |
3p25.3 |
Monoubiquitinated ID complex recruits the downstream repair proteins and facilitates repair of DNA ICLs |
|
FANCE |
6p21.31 |
FA core complex; bridge between the FA core complex and FANCD2 |
|
FANCF |
11p14.3 |
FA core complex |
|
FANCG/XRCC9 |
9p13.3 |
FA core complex |
|
FANCI |
15q26.1 |
Monoubiquitinated ID complex recruits the downstream repair proteins and facilitates repair of DNA ICLs |
|
FANCJ/BRIP1 |
17q23.2 |
FA core complex |
|
FANCL |
2p16.1 |
E3 ubiquitin-protein ligase, monoubiquitination of FANCD2 |
|
2 FANCM |
14q21.2 |
FA core complex. Acts by sensing stalled fork by ICLs and recruiting the core complex proteins to the site of ICL |
|
FANCN/PALB2 |
16q12.2 |
Homologous recombination |
|
2 FANCO/RAD51C |
17q22 |
Resolution of D-loop structures through Holliday Junction Intermediates and Homologous DNA Pairing and Strand Exchange. |
|
FANCP/SLX4 |
16p13.3 |
Cooperate with FANCQ-XPF to generate endonucleolytic incisions to unhook the ICL. |
|
FANCQ/XPF |
16p13.12 |
DNA endonuclease, involved in homologous recombination; responsible for 5′ incision to remove ICLs |
|
2 FANCR/RAD51 |
15q15.1 |
Interact with the ssDNA-binding protein RPA, RAD52 homologous pairing and strand transfer of DNA |
|
2 FANCS/BRCA1 |
17q21.31 |
Homologous recombination |
|
FANCT/UBE2T |
1q32.1 |
E2 ubiquitin-conjugating enzyme, associates with FA core complex, catalyzes monoubiquitination of FANCD2 in association with FANCL |
|
FANCU/XRCC2 |
7q36.1 |
Homologous recombination |
|
FANCV/REV7 |
1p36.22 |
Translesion DNA synthesis |
|
FANCW/RFWD3 |
16q23.1 |
RING-Type E3 Ubiquitin Transferase |
- Cytogenetic location was obtained from OMIM. 2 Also called Fanconi anemia-like genes, mainly due to the absence of bone marrow failure in the patients [9][10].
- Cytogenetic location was obtained from OMIM. 2 Also called Fanconi anemia-like genes, mainly due to the absence of bone marrow failure in the patients [2,9,10].
The FA/BRCA pathway is activated for repairing ICLs during the S phase of the cell cycle, when the replisome finds an ICL and two convergent replication forks become stalled [11]. The ICL repair process can be divided into modules of activity of the FA/BRCA pathway [12] (Figure 1).
(1) Lesion recognition. FANCM and its interacting partners, FAAP24, MHF1 and MHF2 [3], detect the lesion on the DNA when two replication forks converge at the vicinity of an ICL [11]. Replisome complexes are unloaded, leaving stalled replication forks with single stranded DNA (ssDNA) regions covered by Replication Protein A (RPA). This leads to an ATR/CHK1 signaling activation to trigger DNA damage checkpoints [13]. The best described scenario for triggering ICL repair implicates the convergence of two replication forks at the ICL site [11], the leading strand on one side of the ICL stops 20–40 nucleotides before the ICL, and then the CMG helicase is removed from the stalled fork, aided by the ubiquitin E3 ligase TRAIP, the p97 ATPase and FANCS/BRCA1 protein; the fork advances to nucleotide 1 with respect to the ICL and waits for the opposite fork to reach the ICL in a similar manner. Once FANCM and its interacting partners are in close proximity to the ICL, their key function is to recruit the members of the next module, the FA core complex, to the chromatin [14].
(2) FA core complex recruitment. The best described function of the FA core complex is as an E3-ubiquitin ligase that is integrated by the proteins FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP100, FAAP20, FAAP24 and FANCT. FANCL has the E3 ubiquitin ligase catalytic activity and FANCT bears the E2 catalytic activity. Three subcomplexes can be recognized: (a) the FANCB–FANCL–FAAP100 (BL100) subcomplex is important for the integration of all components of the FA core complex; (b) the FANCA–FANCG–FAAP20 (AG20) subcomplex is important for the nuclear localization of the entire multimer; and (c) the FANCC–FANCE–FANCF (CEF) subcomplex is in charge of bridging the FA core complex with the members of the module 3 (its target), the FANCI-FANCD2 (ID2) complex [3]. Once assembled, the entire FA core complex exerts its E3-ubiquitin ligase activity to add a ubiquitin group to FANCI at lysine 523 and to FANCD2 at lysine 561.
(3) ID2 complex monoubiquitination. The FANCD2-FANCI heterodimer, frequently called the central complex, is recruited to the stalled fork, where FANCI is tri-phosphorylated by the ATR-kinase [15], stimulating the FA core complex mediated monoubiquitination of both FANCI and FANCD2. The tri-phosphorylation of FANCI also inhibits the deubiquitinase activity of the USP1-UAF1 complex over the ID2 complex until ICL repair and replication are completed [15]. The ubiquitinated ID2 complex protects the replication forks and regulates the activity of the proteins involved in the processing of the ICL, enabling the recruitment of the proteins of the fourth module.
(4) Homologous recombination (HR) by downstream proteins. Proteins acting downstream in the FA/BRCA pathway include BRCA2/FANCD1, BRIP1/FANCJ, PALB2/FANCN, RAD51C/FANCO, RAD51/FANCR, BRCA1/FANCS, XRCC2/FANCU, XPF/FANCQ, SLX4/FANCP, REV7/FANCV and RFWD3/FANCW, all of which are committed to remove the ICL and maintain genomic integrity through various types of DNA repair. The ubiquitinated ID2 complex recruits the FANCP/SLX4 scaffold protein, which in turn coordinates the endonucleolytic activity of FANCQ/XPF. This endonuclease makes DNA incisions on both sides of the ICL and unhooks it [16] [16]. After ICL unhooking, different types of lesions are generated: one of the chromatids is left with a single strand DNA region with the unhooked ICL, and in the sister chromatid a double strand break (DSB) is generated (Figure 1). All these lesions are repaired by different DNA repair pathways that act coordinately in the FA/BRCA pathway.
The single strand region is repaired by translesion synthesis (TLS), through polymerases REV1 and the polymerase ζ complex (REV7/FANCV-REV3), an error-prone polymerase that uses as template the complementary strand with the adduct; while this allows the replication progress, the low fidelity of this polymerase can introduce errors in the nucleotide sequence [17]. This polymerase is necessary for ICL repair as part of the FA/BRCA pathway; indeed, its recruitment to ICL repair intermediates is performed by ubiquitinated PCNA and FA core complex [18]. The unhooked adduct in the opposite strand is repaired by nucleotide excision repair (NER); FANCQ/XPF protein participates in both FA/BRCA and NER pathways, however FA patients with mutations in this gene, do not share the phenotype with Xeroderma Pigmentosum patients, making evident that is a multitask protein [19]. During ICL repair, FANCQ/XPF makes incisions around the ICL and NER polymerase eta (POL η) is recruited by FANCD2. FA proteins FANCM and FANCT have been implicated in the regulation of NER; these data show the crosstalk between FA/BRCA and NER pathways in the ICL repair [9].
The ICL-associated DSB is processed by the proteins downstream of the FA/BRCA pathway (FANCD1/BRCA2, FANCN/PALB2, FANCS/BRCA1, FANCJ/BRIP1, FANCO/RAD51C and FANCR/RAD51); this set of proteins is recruited by ubiquitinated ID2 complex and performs an homology-directed repair, using the recently restored sister chromatid to perform the error-free HR repair. When the lesion is repaired, the cell is able to continue the cell cycle. Interestingly, some of the downstream proteins have also recently been shown to have functions upstream of the ubiquitinated FANCI-FANCD2 complex. For example, deubiquitinated FANCI and FANCS/BRCA1 are involved in the recruitment of the FA core complex [20]; FANCS/BRCA1 is required for positioning FANCD2 at the ICL site, whereas FANCD1/BRCA2 and FANCJ promote the FANCD2 chromatin localization [9]. Importantly, individuals who are heterozygous for PV in genes of Module 4 are at high risk for developing breast and ovarian cancer [2,21].
(5) ID2 complex deubiquitination. After ICLs repair the activity of the replication fork is restarted; the last module includes the deubiquitination of FANCD2/FANCI activated complex, leading to the re-start of the DNA synthesis by the canonical DNA polymerases. The deubiquitination is performed by the USP1-UAF1 complex resulting in the release of the ID2 complex from the chromatin to complete the ICL repair cycle [12].
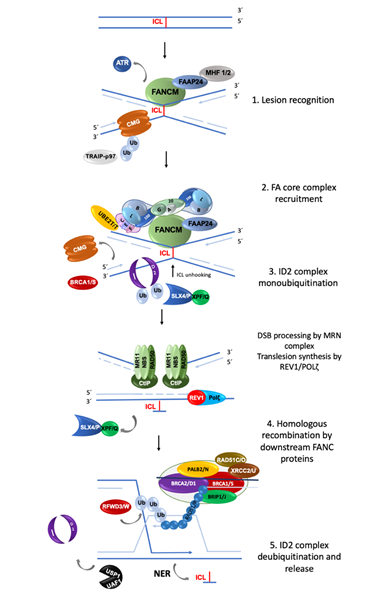
Figure 1. Summary of the activity of FANC proteins in the FA/BRCA pathway. The main function of this pathway is the removal of DNA interstrand crosslink (ICL); 22 FANC proteins participate in: (1) Lesion recognition. FANCM and its partners recognize ICLs during the convergence of two replication forks and promote ATR activation; the CMG helicase complex is unloaded to allow the approach of the leading strands to the ICL. (2) FA core complex recruitment. FANCM and its partners recruit the FA core complex and UBE2T/FANCT (the “upstream” proteins), to exert their E3-ubiquitin ligase activity and monoubiquitinate the FANCI and FANCD2 heterodimer (also known as the “ID2 or central complex”). (3) ID2 complex monoubiquitination. The monoubiquitinated central complex activates the endonucleolytic function of FANCP-SLX4-FANCQ/XPF resulting in the unhooking of the ICL from one of the DNA strands and the generation of a DSB. Both DNA ends of the DSB are processed by the MRN/CtIP complex to form a 3′ overhang. In the opposite strand, the unhooked ICL has now become an adduct; to bypass it, the REV1-polymerase ζ complex (including FANCV protein) performs translesion synthesis of the new strand. (4) The processed DSB is repaired by homologous recombination. The “downstream” FANCD1/BRCA2, FANCN/PALB2, FANCS/BRCA1, FANCJ/BRIP1, FANCO/RAD51C and FANCR/RAD51 proteins coordinate to coat the processed DNA strand of the DSB with RAD51/FANCR and paralogs RAD51C/FANCO, to invade the newly polymerase ζ-synthesized double strand of its sister chromatid, using it as a template to recover the original nucleotide sequence. (5) The cycle finishes with deubiquitination and unloading of the ID2 complex by USP1-UAF1 and the removal of the ICL-adduct by NER (nucleotide excision repair) pathway[21] [9,12,22–24][22][23][24].
2.2. Repair of Double Strand Breaks
DSBs are generated as a byproduct during the processing of ICLs by the FA/BRCA pathway. DSBs are considered one of the most toxic lesions for cells; misrepair may originate mutations and chromosomal abnormalities that may lead to cell death or tumorigenesis, therefore the accurate repair of this type of lesion is essential to maintain genomic stability and cell viability.
Several factors influence the processing and repair of DSBs including the phase of the cell cycle in which the damage occurs, their origin (associated to replication fork stalling or replication-independent) and the number of DSB events in the same cell, among others. The two major repair mechanisms for DSBs are HR and non-homologous end joining (NHEJ); for the latter, a canonical NHEJ (cNHEJ), and an alternative pathway, also called microhomology-mediated end joining (MMEJ), have been described (Table 2). HR uses an intact homologous sequence as a template for the repair of DSBs. For this reason, HR is carried out during the post-replicative period of the cell cycle, which includes the S and G2 phases, when a sister chromatid is available; the free DNA ends have to search for the homologous sequences, thus requiring extensive DNA resection and processing. On the contrary, the ligation of non-homologous ends performed by NHEJ requires minimal or no sequence homology and allows ligation of DNA ends with minimal processing.
In normal cells, NHEJ efficiently joins the correct DNA ends of a DSBs, without the formation of chromosomal aberrations, although the original DNA sequences flanking the DSB may not be exactly restored, due to small losses of nucleotides that occur during the DNA end-processing that is needed for successful NHEJ. However, if multiple DSBs occur simultaneously, the activity of NHEJ, which can be independent of template and homology, may lead to ligation of wrong DNA ends generating gross chromosomal rearrangements [22].
Table 2. Characteristics of the major double-strand break repair mechanisms [22,25,26][25][26].
|
|
Non-Homologous End-Joining |
Microhomology Mediated End-Joining |
Homologous Recombination |
|
Timing |
Fast |
Fast |
Slow |
|
Template dependence |
Independent |
Independent |
Dependent |
|
Homology usage |
0–4 bp |
2–20 bp |
>100 bp |
|
End resection |
no |
yes |
yes |
|
Cell cycle phase |
G1, S, G2 |
G1, S, G2 |
S/G2 |
|
Accuracy of repair |
Mostly accurate, error prone |
Frequently error prone |
Highly accurate |
2.2.1. Homologous Recombination
In a normal cell, the FA/BRCA pathway continues “downstream” after the generation of a DSB, using the HR repair pathway to join the DNA ends in an error free manner. HR is restricted to the S and G2 phases of the cell cycle, using the sister chromatid as template to recover the original nucleotide sequence [22]. However, in any phase of the cell cycle, Ku70-Ku80 are abundant proteins that bind the broken DNA ends and protect them [25]. Since DSBs in S/G2 phases are preferentially repaired by HR, the Ku70-Ku80 heterodimer is removed by proteins that process the DNA ends. A first step in this processing is mediated by the MRN complex (MRE11–RAD50–NBS1), which, aided by CtIP (CtBP-interacting protein), introduces an endonucleolytic nick up to 300 bp away of the DSB site [22]. Next, the 3′ to 5′ MRN exonuclease activity extends the nick forming a 3′ overhang. This process finally ends up displacing the Ku70-Ku80 proteins and elicits the entrance of the late DNA end resection proteins EXO1 (exonuclease1) and BLM-DNA2 (bloom syndrome helicase-endonuclease2) [22,27][27]. These proteins facilitate unwinding of the DNA and digestion of the 5′ strand to lengthen the 3′ overhang, which allows the entry of the RPA protein complex, to protect the single stranded DNA and proceed to HR repair by the FANC proteins (Figure 2) [22].
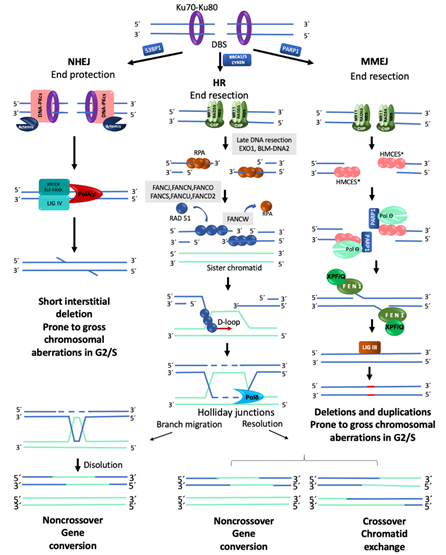
Figure 2. Major mechanisms of double-strand break repair. NHEJ, Non-Homologous End Joining. DNA ends are protected by KU70/KU80, which prevents their end-processing. When the ends are incompatible, a segment of up to four nucleotides is detected, Artemis eliminates the remaining incompatible segments and ligation is carried out by LIGIV-XRCC4. This pathway can repair a DSB without chromosome modification; however, during S/G2 and in the presence of several DSBs, it is considered prone to generate chromosomal alterations. HR, Homologous Recombination. This pathway is only available during the S/G2 phases of the cell cycle since it requires a homologous template; HR is the best choice to maintain sequence fidelity because in general it repairs in an error-free manner. A ssDNA 3′ overhang is produced by the action of the MRN-CtIP complex; it is first covered by RPA proteins which are later replaced by RAD51 to form the nucleoprotein strand that will carry out the invasion of the DNA of the sister chromatid in order to use it as a template to restore the continuity of the original nucleotide sequence. MMEJ, Microhomology-Mediated End Joining. PARP1 prevents KU70/KU80 binding to DNA ends and allows the recruitment of MRN-CtIP to initiate end-resection, creating a short 3′ overhang. This overhang is preferentially covered by HMCES which channels the damage to be repaired by MMEJ instead of HR. PARP1-POLQ search for microhomology of 2–20 bp and align the strands. The resulting flaps are eliminated by XPF/FANCQ-FEN1. Alternatively, POLQ can direct DNA synthesis to add nucleotides to make DNA ends compatible; this end processing generates in situ deletions and duplications. In addition, if this pathway is active during S/G2, and several DSBs coincide in time and space, gross chromosomal aberrations are formed because, similar to NHEJ, they do not require long stretches of homology to ligate the DNA ends [23,26]. * RPA or HMCES (5-hydroxymethylcytosine binding, embryonic stem cell-specific protein) [26,28][28].
The mediators of HR, FANCD1/BRCA2, FANCS/BRCA1, FANCN/PALB2 and FANCJ/BRIP1, act by displacing RPA from the single stranded DNA and loading FANCR/RAD51 and its paralogs FANCO/RAD51C and FANCU/XRCC2 into the ssDNA, which leads to the formation of a nucleoprotein filament with the capacity to invade the sister chromatid and search for homologous sequences that will be used as template to restore the original sequence that was interrupted by the DSB. This nucleofilament assists the base-pairing when the complementary sequences in the sister chromatid have been found (synapsis). Additional FANC proteins mediate the homology search, including FANCW/RFWD3, a ubiquitin E3 ligase that regulates the turnover of RPA by FANCR/RAD51, initially promoted by FANCS/BRCA1. Interestingly, FANCS/BRCA1 has also been shown to promote DNA end resection, RAD51 loading and collaborate in the homology search mediated by FANCR/RAD51, highlighting the multiple roles of FANCS/BRCA1 [22,27] (Figures 2 and 3).
When the synapsis has been stabilized, dissociation of FANCR/RAD51 is required to promote DNA synthesis. A displacement-loop (D-loop) is then formed allowing the engagement of the DNA polymerase δ (Pol δ) [14,23,26,29][29] to incorporate nucleotides and synthesize new DNA. The DNA heteroduplex leads to the formation of Holliday junctions, which are resolved by helicases and endonucleases. The Holliday junctions can be either dissolved or resolved, the first option restores the original sequence by gene conversion (non-crossover), whereas the second can promote sister chromatid exchanges (crossover) or gene conversion (non-crossover), this prevents chromosomal translocations, as expected by an error-free repair [30,31][30][31] (Figure 2). Importantly, HR preferentially uses the sister chromatid as a template due to its perfect homology and close proximity, though the use of the homologous chromosome is also possible, however this alternative is less efficient and can generate regions of homozygosity in the next cell generation.
Once the DSB is repaired, FANCD2-Ub has to be extracted from the lesion; for this to occur, it has to be deubiquitinated by the USP1-UAF1 deubiquitinase complex and p97. This deubiquitination step is the process best-known to contribute to finalize the HR [12[32],32], however all known “downstream” FANC proteins not only have important functions in HR, but some of them also control the start and conclusion of the repair cycle, as well as the DSB repair pathway choice [26].
During the processing of DNA ends, the ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia related) kinases become activated by MRN and RPA, respectively, and regulate additional aspects of the cell’s DNA damage response, including activation of the cell cycle checkpoints.
In patients with FA, the error-free FA/BRCA pathway is not functional, so their cells have to select an alternative error-prone pathway to repair their DNA. The routes they usually use are the NHEJ and the alternative end joining also called microhomology mediated end joining (MMEJ). These error prone pathways are described below.
2.2.2. Non-Homologous End Joining
NHEJ is the dominant pathway for the repair of DSBs in the human cells [25]. Processing of a DSB by NHEJ is notably different from HR. As observed in Table 2, the abundance and availability of its components throughout all the cell cycle and the speed of DSB repair kinetics (15–30 min) explain the dominant role that cNHEJ has in the preservation of genome integrity. The initiation of the classical cNHEJ requires the union of the Ku70/Ku80 heterodimer to the broken DNA ends. This protects the DNA from the exonuclease activity of proteins such as MRN or EXO1.
Ku70/Ku80 is also a platform for the recruitment of other DNA repair proteins, such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and its cofactor with exonuclease activity called Artemis. The Ku70/Ku80-DNA-PKcs complex makes a first synapse between the two DNA ends followed by a second synapse with a closer contact operated by the proteins DNA ligase IV (LIG4), XRCC4/XLF, PAXX, DNA polymerases λ and μ, Aprataxin and PNK-like factor (APLF). These proteins are also in charge of performing DNA end processing by removing some nucleotides from the broken ends to allow the ligation between either blunt DNA ends or DNA ends with a very short resection that leads to small single stranded DNA overhangs (Figure 2). This resection (≤4 pb), although small, can change the information in the damaged site, explaining part of the errors associated with DNA repair by cNHEJ [22,25,26]. The cNHEJ simplicity explains in part its high speed, enabling the re-ligation of DSBs shortly after they were formed. High speed in cNHEJ partially compensates for the lack of homology use. When a single DSB occurs, the proximity and topology of the two original DNA ends increases the probability of its re-ligation; however, when more than two DNA ends coexist, the template-independent ligation that characterizes NHEJ increases the probability of generating gross chromosomal rearrangements [33].
2.2.3. Alternative End Joining (Microhomology Mediated End Joining)
When HR or cNHEJ are unavailable, the cell can use MMEJ; the distinctive characteristic of this repair pathway is the use of very short homologous sequences (2–20 bp) to elicit the re-joining of the two DNA ends. MMEJ is an error prone pathway with characteristics similar to the cNHEJ but that also includes DNA end processing. MMEJ requires PARP1 (poly(ADP-ribose) polymerase 1), a protein that competes with KU for the DNA ends generated by a break [34], the binding of PARP1 facilitates the recruitment of DNA polymerase θ (Pol θ) to DSBs [35]. For DNA end resection, MMEJ recurs to CtIP and the MRN complex to create a 15–100-nucleotide 3′ overhang. This ssDNA 3′ overhang coated by proteins RPA or HMCES (5-hydroxymethylcytosine binding, embryonic stem cell-specific protein) [28] makes a short displacement until it finds a microhomology region of up to 20 bp, leaving a 5′ flap. At this point, HMCES are unloaded through the Pol θ-associated helicase activity, the displaced 5′ ssDNA flaps are removed by FEN1 (flap endonuclease 1) and DNA ligase I or DNA ligase III closes the dent [36] (Figure 2).
When no homology is found, the polymerase activity of Pol θ is turned on to add nucleotides and provide the necessary microhomology to stabilize the junction between the two free DNA ends; either because it removes nucleotides to match existing microhomology regions in DNA ends or because it inserts nucleotides to create microhomology regions, MMEJ is prone to introduce deletions and duplications [26,28]. Pol θ is enhanced in HR defective cells, suggesting that this type of repair may act when the DNA ends cannot be repaired by the cNHEJ [26]. The high levels of chromosome translocations observed in cNHEJ mutants that use MMEJ indicate that this process tends to bind together non-homologous segments and therefore produces structural chromosomal aberrations (SCA) [28,36][36]. Apparently, MMEJ functions as a backup route when HR and cNHEJ fail to resolve the DSBs present in the cell. [33].
2.2.4. DSB Repair Pathway Choice
Some calculations suggest that the cell responds to even a single DSB by activating cell cycle checkpoints; it has been estimated that the integrity of the genome and cell survival is put at risk when several DSBs (~10) are simultaneously induced [37]. Therefore, the choice of the DNA repair pathway to maximize the efficiency to preserve genome integrity is critical for the survival of any cell. Although several pathways and sub-pathways have been implicated in the repair of DSBs, here we only consider the three main pathways: HR, NHEJ and MMEJ.
The phase of the cell cycle in which a DSB occurs is one of the most important and defining characteristics for DNA repair pathway choice. HR repair is not active in G1 because sister chromatids are not available, therefore DSBs appearing in this phase will be channeled to NHEJ or MMEJ. When a DSB occurs in S/G2 phases, HR is the preferred pathway for its repair since it is the best way to preserve the integrity of the DNA sequence, even if end joining mechanisms are active. Although the exact mechanism behind the DSB repair pathway choice remains elusive, proteins driving the initial steps of DSB processing are the candidates to determine the selection of the best pathway [26].
Nucleolytic processing of the DNA-ends is a critical step during DSB repair pathway choice (Figure 2). During the G1 phase of the cell cycle, an active suppression over end resection machinery, specifically MRN, is performed by the 53BP1 protein and the shieldin complex, thus restricting HR to S/G2 phases, and leaving the Ku70/Ku80 heterodimer without competitors during its DNA end-protection activity. Through suppression of DNA end processing, 53BP1 favors NHEJ. During the postreplicative phases (S and G2), the FANCS/BRCA1 protein, in collaboration with MRN and CtIP, antagonizes 53BP1 and promotes the essential DNA end resection step for both HR and MMEJ [38]. Posttranslational modifications (PTM) of histones have been shown to be important for chromatin localization of 53BP1: it requires the combined interaction of its Tudor domain to H4K20me2 and the H2AK15ub through a C-terminal ubiquitin-interacting motif [39]. The FA/BRCA pathway, via FANCD2, restrains the accumulation of 53BP1 by regulating the activity of TIP60, an acetylase of histone H4 that increases the presence of H4K16ac and H2AK15ac in the site of DNA damage hindering access to the post-translational modification required to maintain 53BP1 in the chromatin. Failure of the FA/BRCA pathway leads to 53BP1 accumulation favoring NHEJ, leading to chromosomal aberrations [12].[40][41][42][43][44][45]
The HR, NHEJ and MMEJ pathways are all active during S/G2; therefore, to channel DSBs repair to HR during this cell cycle phase, it is necessary to repress the activity of cNHEJ. Recently, a specific inhibitor of NHEJ in the post replicative phase has been proposed, CYREN (cell cycle regulation of NHEJ), which binds the Ku70/Ku80 complex and regulates the DNA repair pathway choice by inhibiting NHEJ and promoting HR when a sister chromatid is available to allow error free recombination [40].
2.3. Double Strand Breaks as the Substrate for Chromosomal Aberrations
The most common CAs observed in metaphase spreads from patients with FA are chromatid or isochromatid breaks, deletions, duplications, fragments and gross chromosomal aberrations, such as translocations, dicentric chromosomes, radial figures and other complex rearrangements (Figure 3) [41][46]. The formation of all of these SCAs involves breaking and rejoining of DNA molecules, therefore DSBs are considered to be the origin of SCA [42][47].
The heterogeneous clinical phenotype observed in the patients with FA contrasts with their highly constant cellular and cytogenetic phenotype. The homogeneous cellular phenotype observed in FA indicates that failure in any stage of the FA/BRCA pathway results in the incapacity for repairing ICLs and DSBs in an error-free manner. Misrepaired DSBs in particular, which arise after initial ICL processing, are the main source of chromosomal aberrations (CA) present in FA cells, and this sensitivity has been critical for the diagnosis of FA, which is largely based on the detection of CA observed in cell cultures treated with Diepoxybutane (DEB) and mitomycin C (MMC) [43][48].
2.3.1. Non-Rejoined Structural Chromosomal Aberrations: Breaks
In FA cells, chromosome breakage is the result of initiated but unfinished ICL repair. Most of the breaks in FA cells are of the chromatid type (Figure 3b,c), indicating that they were formed during the post-replicative period of the cell cycle, and therefore only one chromatid is affected, even though the chromosome is already composed by two sister chromatids. Generally, when a break of the chromosomal type (both chromatids are broken) is detected, it can be inferred that a DSB occurring during the G1 phase is the cause. Nonetheless, the FA pathway operates in the S/G2 phases, therefore it is more likely that chromosomal breaks are the result of two very close DSBs, one in each chromatid, and can be considered isochromatid breaks. Isochromatid breaks are less common than chromatid breaks when metaphase spreads from FA samples are analyzed.
Since the FA/BRCA pathway is not functional in FA cells, the presence of chromosome breaks, when FA cells are treated with ICL inducing agents, suggests that endonucleases, alternative to the canonical FA/BRCA pathway, unhook the ICL and generate a DSB. This DSB however is not channeled to HR by the downstream modules of the FA/BRCA pathway and might remain unrepaired; when a cell reaches metaphase, the sites of these unrepaired DSBs can be visualized as chromatid breaks. Of note, the piece of broken chromatid usually remains adjacent to its chromosome due to the mitotic chromatin structure and the cohesin proteins that hold together the sister chromatids and prevent their separation until anaphase [44] (Figure 3c).
2.3.2. Rejoined Structural Chromosome Aberrations
In FA cells, the presence of translocations, dicentrics and radial figures makes evident the relevance of FA/BRCA pathway in the protection against SCAs, since all of these aberrations originate by ligation of two or multiple broken DNA ends with little or absent homology. If the end joining pathways cNHEJ and MMEJ operate during S/G2 phases of the cell cycle, when the replicated chromosome is composed by two sister chromatids, the joining of one of these chromatids with a non-sister chromatid from a different chromosome (homologous or non-homologous) will lead to SCA formation (Figure 3).
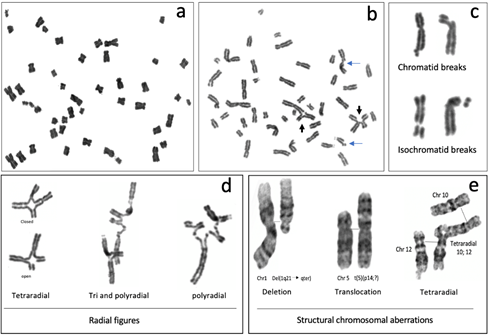
Figure 3. Representative metaphases and common structural chromosome aberrations observed in FA cells cultured with 0.1 µg/mL DEB: (a–d) peripheral blood lymphocytes from a patient with FA and (e) from a patient derived, FANCA mutated VU817 lymphoblastoid cell line. (a) Endoreduplication. (b) Metaphase with structural chromosomal aberrations; blue arrows show chromatid breaks and black arrows show radial figures. (c) Breaks. The images show how the broken fragments are kept very close to the chromosome that originated them because there is a cohesion of chromatids in metaphase. (d) Radial figures. In the first column can be observed that the tetraradial can be closed when the four DNA ends of the two non-homologous chromosomes are rejoined or open when only two of the four DNA ends were rejoined. (e) GTG banded chromosomes reveal other gross structural chromosomal aberrations that may be found in FA cells, such as deletions and translocations. In the radial figure, normal chromosomes 10 and 12 are aligned with the tetraradial, to highlight the trajectory of the rearrangement.
Radial figures are formed when at least two DSBs from non-sister chromatids are joined together. In these two DSBs, four DNA ends are available; therefore, if the four DNA ends are joined by an error prone DNA repair machinery, a closed tetraradial can be generated. However, an open tetraradial figure will be originated if only two DNA ends are rejoined (Figure 3d). A triradial figure has the pre-requisite of three DSBs, one of them in a chromatid of the receptor chromosome and two more (of the isochromatid break type), in the second chromosome to join one chromatid of the receptor chromosome; in this way, polyradial figures require several DSBs for their formation (Figure 3d). In FA cells, radial figures form between non-homologous chromosomes (Figure 3e) [45][42], both spontaneously and induced by MMC or DEB. When proteins in both the HR and cNHEJ pathway are inactivated, an increase in the frequency of radial figures can be observed [46][43], suggesting that the MMEJ pathway highly contributes to its generation.
Translocations, dicentric chromosomes and chromosome deletions may be directly originated during the abnormal processing of ICLs or arise as a consequence of the extremely abnormal segregation that radial figures undergo during mitosis. Depending on the type of radial chromosome, the transition through anaphase will result in the segregation to the daughter cells of translocated chromosomes, dicentric chromosomes, acentric fragments and deleted chromosomes (Figure 4). Importantly, large numbers of cells can succumb to cell death by the accumulation of gross genomic imbalances, i.e., radial figures can lead to anaphase bridges and mitosis blockage, or chromosome fragments can lead to micronuclei formation after cytokinesis, which can create a vicious circle of CIN that can eventually result in the emergence of neoplastic clones. Of note, each cell with at least one radial figure can give rise to four different daughter cells, carrying different non-clonal chromosomal alterations. This makes clear that a cell with several SCA will generate daughter cells with karyotypes different from the progenitor cell, generating a wide diversity of genotypes (Figure 4).
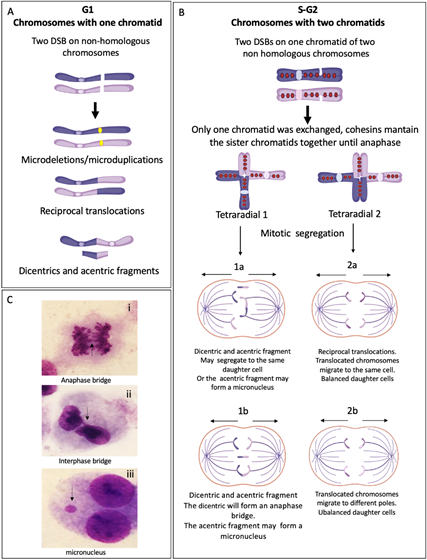
Figure 4. End joining repair pathways outcomes when more than one DSB is present in the same space-time and the rejoining is between non-homologous chromosomes. (A) G1 chromosomes have only one chromatid. When two chromosomes have DSBs, repair by NHEJ and MMEJ can perform the reunion of the two original chromosomal fragments, without error or leaving microdeletions or microduplications. If two fragments from different chromosomes are joined, translocations or dicentrics + acentric fragments are generated. (B) S/G2 chromosomes have two sister chromatids linking each other by cohesins. If only one sister chromatid has a DSB, the interchange of segments during repair generates gross structural aberrations such as translocations or radial figures, which may have several configurations depending on the rejoined fragments. Here, we show two possible tetraradial figures with different outcomes after segregation. (1) The segregation of a closed tetraradial with two DNA ends rejoining chromatids with centromere: a dicentric. (1a) In this type of segregation, both normal chromosomes segregate in a daughter cell and the dicentric chromosome moves together with the acentric fragment to the second daughter cell. (1b) The normal chromosomes segregate each to a different daughter cell, the dicentric is attached to both centrosomes of the mitotic spindle, and an anaphase bridge is formed, with a high probability of breaking at some point, generating chromosomes with deletion or translocation. In any type of segregation, the acentric fragment can form a micronucleus. (2) Segregation of a tetraradial with two DNA ends rejoining segments without centromere. (2a) Both chromatids with a translocated centromere segregate to the same pole, the result is one daughter cell with balanced translocation and one normal daughter cell. (2b) The translocated chromosomes segregate to different daughter cells, both will have unbalanced translocations. (C) Cells from a FA patient showing: (i) anaphase bridge; (ii) interphase bridge, resulting of the segregation failure of a dicentric; and (iii) micronucleus, frequently formed by an acentric fragment that could not join the mitotic spindle.
2.3.3. Other Chromosome Aberrations
All of these SCA can be accompanied by numerical alterations, such as aneuploidies (gains or losses of whole chromosomes) and polyploidization. In metaphase spreads of patients with FA, it is relatively common to find tetraploid cells and mitotic figures with endorreduplicated chromosomes, with four instead of two chromatids. FA cells are also known to have alterations in the duration of the cell cycle phases (explained below) or in the transition from one phase into another. These might provoke new DNA replication cycles in the absence of mitosis and cytokinesis, leading to endorreduplicated chromosomes in the next mitosis (Figure 3a) [47][44]
2.4. Chromosome Aberrations for the Diagnosis of Fanconi anemia
Presence of SCAs is a hallmark of the FA cellular phenotype, therefore the analysis of the number and type of SCA is used in the diagnosis of FA. An approximate 10-fold increase in the DEB-induced frequency of SCA and the presence of radial figures are indicative of FA (Figure 5). In some patients, the diagnostic chromosome breakage test for ruling out FA might turn out to be inconclusive due to the presence of a subpopulation of cells that are not sensitive to DEB or MMC and behave as normal cells. In these cases, the presence of a revertant cell line giving rise to mosaicism should be sought. Mosaicism in the context of FA refers to the existence, in a single patient, of two hematopoietic cell populations, one sensitive and one resistant to ICL-inducing agents. Mosaicism appears due to the reversion of one of the original germline PV causing FA. It is calculated to be present in up to 20% of patients with FA and can have multiple origins, including gene conversion, back mutation, second-site mutation, among others. There is no standard methodology for the diagnosis of mosaicism, however a patient is generally considered to have hematopoietic mosaicism when a sub-population of his/her lymphocytes displays DEB resistance, while their fibroblast show DEB sensitivity [48][45]. The presence of mosaicism has clinical implications, if the reversion occurs early in the primitive hematopoietic stem cells it might lead to increased blood cell counts, improved aplastic anemia, as well as a reduction in the incidence of bone marrow failure and hematologic neoplasia [48][45].
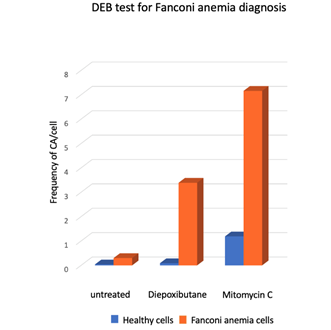
Figure 5. Response of lymphocytes from FA patients (n = 18) and healthy subjects (n = 117) to treatment with ICL inducing agents, Diepoxybutane [0.1 µg/mL] and mitomycin C [40 ng/mL]. Gross chromosomal aberrations such as radial figures, translocations, deletions and duplications were commonly observed in FA cells [49] Although the two challenge agents are effective, the use of DEB for diagnosis is preferred, because the results are less variable and the difference between non-FA vs. FA cells is generally clearer.
References
- Wegman-Ostrosky, T.; Savage, S.A. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. 2017.
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015.
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017.
- Nakamura, J.; Nakamura, M. DNA-protein crosslink formation by endogenous aldehydes and AP sites. DNA Repair (Amst). 2020.
- Schärer, O.D. DNA interstrand crosslinks: Natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem 2005.
- Rosado, I. V.; Langevin, F.; Crossan, G.P.; Takata, M.; Patel, K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011.
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018.
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019.
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016.
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; D’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surrallés, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018.
- Zhang, J.; Dewar, J.M.; Budzowska, M.; Motnenko, A.; Cohn, M.A.; Walter, J.C. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 2015.
- Renaudin, X.; Rosselli, F. The FANC/BRCA Pathway Releases Replication Blockades by Eliminating DNA Interstrand Cross-Links. Genes (Basel). 2020.
- Collis, S.J.; Ciccia, A.; Deans, A.J.; Hořejší, Z.; Martin, J.S.; Maslen, S.L.; Skehel, J.M.; Elledge, S.J.; West, S.C.; Boulton, S.J. FANCM and FAAP24 Function in ATR-Mediated Checkpoint Signaling Independently of the Fanconi Anemia Core Complex. Mol. Cell 2008.
- Kim, J.M.; Kee, Y.; Gurtan, A.; D’Andrea, A.D. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 2008.
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-Mediated FANCI Phosphorylation Regulates Both Ubiquitination and Deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020.
- Zhang, J.; Walter, J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst). 2014.
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1–pol ζ complex during bypass of a DNA interstrand cross‐link . EMBO J. 2015.
- Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020.
- Marín, M.; Ramírez, M.J.; Carmona, M.A.; Jia, N.; Ogi, T.; Bogliolo, M.; Surrallés, J. Functional comparison of XPF missense mutations associated to multiple DNA repair disorders. Genes (Basel). 2019.
- Castella, M.; Jacquemont, C.; Thompson, E.L.; Yeo, J.E.; Cheung, R.S.; Huang, J.W.; Sobeck, A.; Hendrickson, E.A.; Taniguchi, T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet. 2015.
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019.
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 2018.
- Rageul, J.; Kim, H. Fanconi anemia and the underlying causes of genomic instability. Environ. Mol. Mutagen. 2020.
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.; Chorev, D.S.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019.
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019.
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genomics, Proteomics Bioinforma. 2016.
- Shukla, V.; Halabelian, L.; Balagere, S.; Samaniego-Castruita, D.; Feldman, D.E.; Arrowsmith, C.H.; Rao, A.; Aravind, L. HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Mol. Cell 2020.
- Kim, Y.; Lach, F.P.; Desetty, R.; Hanenberg, H.; Auerbach, A.D.; Smogorzewska, A. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011.
- Liu, Y.; West, S.C. Happy Hollidays: 40th Anniversary of the Holliday junction. Nat. Rev. Mol. Cell Biol. 2004.
- Colavito, S.; Prakash, R.; Sung, P. Promotion and regulation of homologous recombination by DNA helicases. Methods 2010.
- Cohn, M.A.; Kee, Y.; Haas, W.; Gygi, S.P.; D’Andrea, A.D. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 2009.
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. - Genet. Toxicol. Environ. Mutagen. 2015.
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006.
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 2018.
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017.
- Löbrich, M.; Jeggo, P.A. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 2007.
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009.
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.C.; Kitevski-Leblanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013.
- Arnoult, N.; Correia, A.; #1, M.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA Repair pathway choice in S/G2 by the NHEJ inhibitor CYREN Europe PMC Funders Group. Nature 2017.
- Srinivasan, M.; Fumasoni, M.; Petela, N.J.; Murray, A.; Nasmyth, K.A. Cohesion is established during dna replication utilising chromosome associated cohesin rings as well as those loaded de novo onto nascent dnas. Elife 2020.
- Newell, A.E.H.; Akkari, Y.M.N.; Torimaru, Y.; Rosenthal, A.; Reifsteck, C.A.; Cox, B.; Grompe, M.; Olson, S.B. Interstrand crosslink-induced radials form between non-homologous chromosomes, but are absent in sex chromosomes. DNA Repair (Amst). 2004.
- Hanlon Newell, A.E.; Hemphill, A.; Akkari, Y.M.N.; Hejna, J.; Moses, R.E.; Olson, S.B. Loss of homologous recombination or non-homologous end-joining leads to radial formation following DNA interstrand crosslink damage. Cytogenet. Genome Res. 2008.
- Kubbies, M.; Schindler, D.; Hoehn, H.; Schinzel, A.; Rabinovitch, P.S. Endogenous blockage and delay of the chromosome cycle despite normal recruitment and growth phase explain poor proliferation and frequent endomitosis in Fanconi anemia cells. Am. J. Hum. Genet. 1985.
- Nicoletti, E.; Rao, G.; Bueren, J.A.; Río, P.; Navarro, S.; Surrallés, J.; Choi, G.; Schwartz, J.D. Mosaicism in Fanconi anemia: concise review and evaluation of published cases with focus on clinical course of blood count normalization. Ann. Hematol. 2020.
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10.
- Yu, V.P.C.C.; Koehler, M.; Steinlein, C.; Schmid, M.; Hanakahi, L.A.; Van Gool, A.J.; West, S.C.; Venkitaraman, A.R. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000.
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185.
- Esmer, C.; Sánchez, S.; Ramos, S.; Molina, B.; Frias, S.; Carnevale, A. DEB Test for Fanconi Anemia Detection in Patients with Atypical Phenotypes. Am. J. Med. Genet. 2004.
- Sedlackova, H.; Rask, M.B.; Gupta, R.; Choudhary, C.; Somyajit, K.; Lukas, J. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature 2020.