The effects of airway inflammation on airway smooth muscle (ASM) are mediated by pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα).
- Airway Inflammation,airway smooth muscle,TNFα
1. Introduction
Airway inflammation underlies a number of pathological conditions such as asthma, chronic obstructive pulmonary disease (COPD), chronic bronchitis, COVID-19 and various cough syndromes. With the COVID-19 pandemic, we are keenly aware of the detrimental impact of airway inflammation and acute respiratory distress syndrome (ARDS). Even before COVID-19, ARDS affected as many as 246,000 people in the USA each year (based on an incidence rate of ~75 per 100,000), with influenza and the common cold affecting many more. The effects of acute airway inflammation are mediated by pro-inflammatory cytokines (e.g., tumor necrosis factor alpha, TNFα) leading to increased contractile protein expression, increased force generation and increased ATP consumption in airway smooth muscle (ASM) [1][2][1,2]. In humans, the increase in ATP consumption in ASM is matched by increased mitochondrial O2 consumption at the expense of increased reactive oxygen species (ROS) formation (oxidative stress) [3]. The subsequent oxidative stress results in an accumulation of unfolded proteins in the endoplasmic reticulum (ER) and in mitochondria triggering unfolded protein responses in both organelles. In a recently published study, we demonstrated that TNFα selectively activates the inositol-requiring enzyme 1 alpha (pIRE1α) mediated ER stress pathway in human ASM, which induces alternative splicing of mRNA for the transcription factor X-box binding protein 1 (XBP1s) [3]. In a follow-up study, we showed that 24 h TNFα exposure increases the expression of peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC1α) in human ASM cells, thereby promoting mitochondrial biogenesis and an increase in mitochondrial volume density [4]. The TNFα induced increase in PGC1α and mitochondrial remodeling could be downstream to either ER stress or the mitochondrial unfolded protein response (mtUPR) [5][6][5,6]. In addition to playing a key role in mitochondrial biogenesis, PGC1α also activates the Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)/Parkin mitophagy pathway, which is involved in the ubiquitination of the mitochondrial fusion protein Mfn2 [7][8][7,8]. Accordingly, we found that acute exposure to TNFα also results in a decrease in mitofusin 2 (Mfn2) protein levels in human ASM cells while expression of dynamin-related protein-1 (Drp1) increases, resulting in mitochondrial fragmentation [4][9][10][4,9,10]. In addition to promoting mitochondrial fusion, Mfn2 also plays an essential role in tethering mitochondria to the ER, thereby establishing the proximity of mitochondria to ER Ca2+ release sites that represent microdomains of higher cytosolic Ca2+ concentrations ([Ca2+]cyt “hotspots”) [9][10][11][12][9,10,11,12]. In another study, we reported that acute TNFα exposure disrupts the tethering of mitochondria to the ER and reduced mitochondrial Ca2+ influx through the mitochondrial Ca2+ uniporter (MCU) [13]. This is consistent with other studies showing that close proximity of mitochondria to the ER is necessary to open the MCU and thereby promote mitochondrial Ca2+ influx [9][10][11][12][9,10,11,12]. In this review article, we provide a conceptual framework for a homeostatic response triggered by activation of the pIRE1α/XBP1s ER stress pathway, which serves to mitigate the impact of TNFα-induced ER stress by promoting mitochondrial biogenesis, reducing mitochondrial Ca2+ influx and thereby limiting ROS formation (Figure 1). Components of this homeostatic response include a pIRE1α/XBP1s-mediated increase in PGC1α and an increase in PINK1/Parkin leading to: (a) a reduction in Mfn2 and restricted tethering of mitochondria to the ER, and (b) mitochondrial biogenesis leading to an increase in mitochondrial volume density, which reduces O2 consumption and ROS formation per mitochondrion, while still meet the increased ATP demand of force generation.
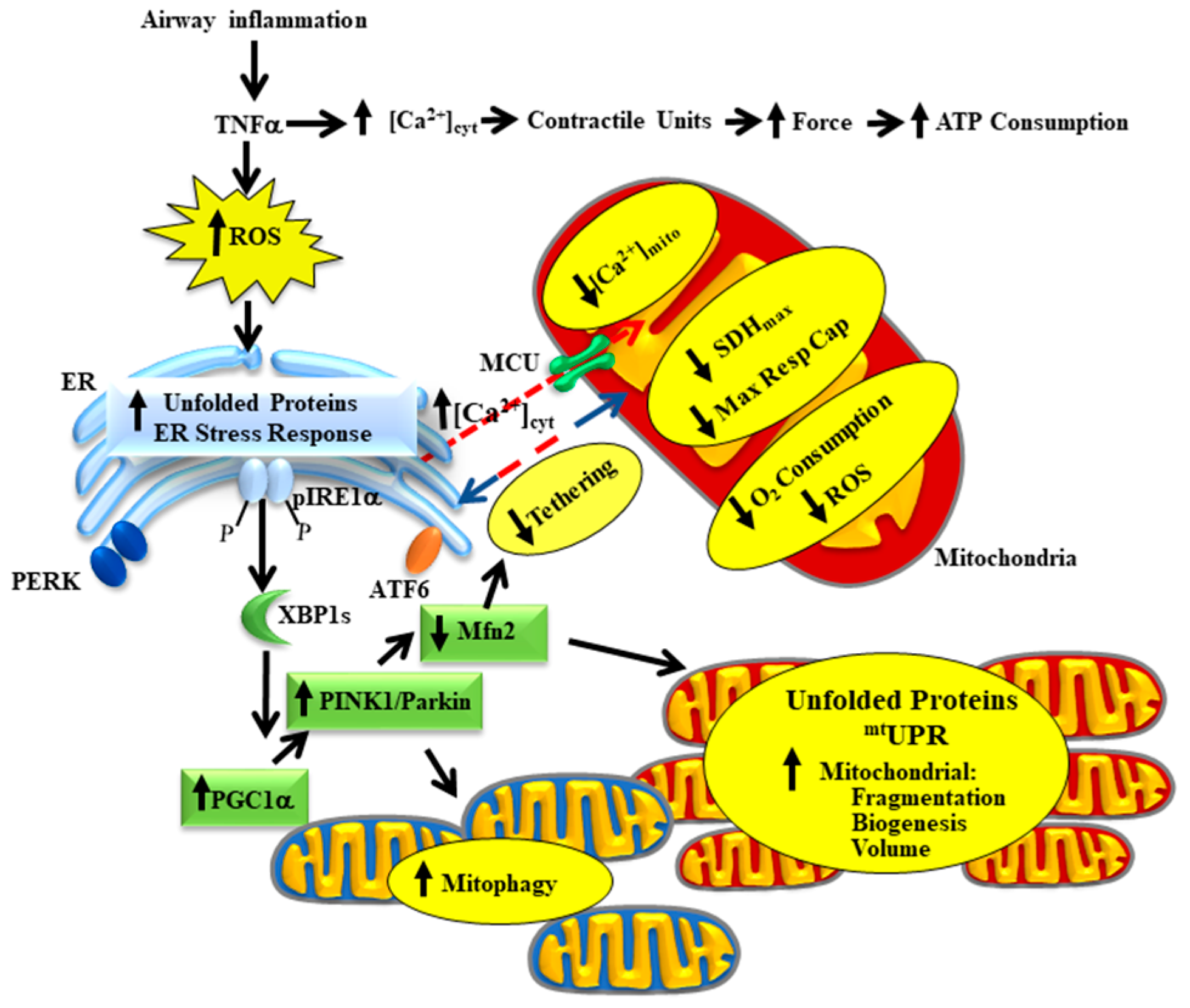
2. The Homeostatic Response to TNFα in Asthmatic Human ASM
Asthma is a chronic inflammatory condition of ASM, characterized by airway inflammation, airway hyper-contractility and airway remodeling (ASM proliferation) [14][15][16][17][18][69,70,71,72,73]. Surprisingly, very few studies on asthma have dealt with the crosstalk between mitochondrial oxidative stress and ER stress response and its impact on the inflammation in human ASM to date. An increased ROS generation is reported in asthmatic patients [14][19][20][69,74,75], which can trigger the ER stress in human ASM. Some studies have demonstrated that the activation of ER stress response in airway epithelial cells or immune cells is enhanced in asthma [21][22][23][19,20,21] but did not specifically explore the contribution of any of the ER stress sensor activation in this context. However, to date, no study has explored the impact of ER stress and its marker activation in human asthmatic ASM. As mentioned above, pro-inflammatory cytokine TNFα selectively induces the pIRE1α/XBP1s ER stress pathway in non-asthmatic ASM [3], and the effect of inflammation is not explored in asthmatic ASM. It is not known whether TNFα mediated ER stress plays a similar homeostatic role maintaining the dynamicity and activity of mitochondria in asthmatic conditions or if the effect is blunted. Additionally, the impact of any other proinflammatory cytokines other than TNFα in asthma is also a question to be answered. Previous studies have shown that in asthmatic ASM, mitochondrial biogenesis is increased [24][25][67,68], and we can speculate that it can be positively correlated with mitochondrial volume density, however, it has not been reported so far. We have reported an increase in mitochondrial fragmentation in human ASM of moderate asthmatics associated with an increase in mitochondrial fission protein Drp1 and a decrease in fusion protein Mfn2 expression in asthma, resulting in more fragmented mitochondrial networks [26][48]. Considering all the experimental evidence, further illustrating studies are needed to understand the impact of inflammation on ER stress and mitochondrial crosstalk in asthma and also to find some novel therapeutic target(s).
3. Conclusions
With the recent emergence of the COVID-19 pandemic, we became concerned with the pathological significance of acute airway inflammation, which is mediated by pro-inflammatory cytokines (e.g., TNFα). TNFα plays a major role in force generation and ATP consumption by ASM during airway inflammation and induces mitochondrial oxidative phosphorylation to match the increased ATP demand. As a result, ROS generation and oxidative stress is increased, which can be deleterious to the system. Simultaneously, as a homeostatic response, TNFα selectively activates the pIRE1α/XBP1s mediated ER stress pathway in ASM and increases mitochondrial biogenesis and mitochondrial volume density and reduces O2 consumption and ROS formation by individual mitochondrion and protects ASM cells from the negative impact of inflammation-induced ROS formation while still meeting the ATP demand. In our belief, a failure in this homeostatic mechanism can lead to increased ROS formation, thereby exacerbating cell death.