Liquid–liquid phase separation (LLPS) is a thermodynamically-driven, reversible phenomenon consisting in de-mixing into two distinct liquid phases, with different solute concentrations.
- intrinsically disordered proteins
- viruses
- liquid condensates
- liquid-liquid phase separation
1. General Concepts of Phase Separation
A physically homogeneous system consists of a single phase and is said condensed when formed by a liquid or a solid. Miscible components can exist in homogeneous solutions or undergo de-mixing, depending on the interactions among solute and solvent molecules. Phase separation can become energetically favored, in spite of the entropic loss entailed by the formation of a two-phase system [1][2][3][99,100,101]. Among possible transformations of a chemically homogeneous system, LLPS is a thermodynamically-driven, reversible phenomenon consisting in de-mixing into two distinct liquid phases, with different solute concentrations [4][5][102,103]. The equilibrium between mixing and de-mixing is strongly dependent on the component concentrations, besides temperature, pressure, pH, crowding agents, etc.
We underline that phase separation of a liquid system does not necessarily imply a phase transition (i.e., a state change from liquid to solid), although the latter can take place during the “maturation” of liquid biological condensates. A phase diagram can be constructed by systematically screening, for instance, temperature and concentration, to determine the region in which phase separation occurs (Figure 1Figure 3). The phase diagram is characterized by a “binodal” (coexistence) curve, which encloses the whole region where phase separation is thermodynamically accessible. A “spinodal” curve encloses an inner region where the single-phase mixture is unstable and phase separation spontaneously occurs, in the absence of nucleation and perturbation, being limited by molecule diffusion and giving rise to two components with similar fractional volume and typical intertwined structures (“spinodal decomposition”). In the region between the two curves, phase separation requires the formation of condensation seeds, through a nucleation step that limits the rate of the de-mixing process. Here, if phase separation does not occur, the lone phase representing the system reaches “super saturation”. This is a “metastable” condition, which can originate a two-phase system even in response to subtle perturbations [6][7][8][9][104,105,106,107].
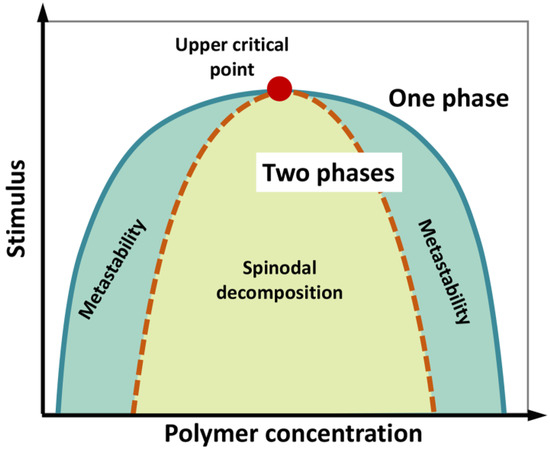
Figure 13. Schematic phase diagram of a colloidal system (e.g., a polymer in water) displaying an upper critical point (filled red point), above which no de-mixing occurs. According to polymer concentration and external stimuli (temperature, pH, ionic strength, etc.), the system consists in a well-mixed, single phase, or two separated phases. The diagram illustrates the coexistence or binodal curve (green) and the spinodal curve (dotted brown). The region in between the binodal and spinodal curves (light-green region) can correspond to metastable, supersaturated solutions. In the region enclosed by the spinodal curve, single-phase mixture is unstable and phase separation (“spinodal decomposition”) spontaneously occurs, in the absence of nucleation, being limited by molecule diffusion. Figure adapted from [9][107].
In the regime of phase separation, liquid droplets can fuse and coalesce, undergoing Ostwald ripening (i.e., larger droplets grow at the expense of smaller ones), and dripping, according to their viscosity and surface tension [10][108]. A “phase behavior” can be easily figured out for compounds of different chemical nature (hydrophobic/hydrophilic), such as oily substances and water, which are notoriously immiscible [4][102]. Immiscible substances differ in their partition coefficient (K), which is calculated as the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium (typically, octanol and water to obtain Kow). The greater the difference between Kow of two molecules, the lower the concentration limit (i.e., solubility). As an example, very small amounts of aniline (Kow = 1.076 Log L/Kg) are completely miscible in water at any temperature below 441 K. As aniline concentration increases, the two components separate into two liquid phases [11][109]. Other than temperature and concentration, several factors, including pH, pressure and salts, influence the phase behavior of solution systems.
2. Phase Separation in Protein Systems
Phase separation has recently emerged as a ubiquitous principle of cellular organization, underlying many biological processes, the list of which is growing rapidly [12][13][14][15][16][17][18][19][89,92,95,96,110,111,112,113] (Figure 2). According to the PubMed database, almost 3000 scientific articles have contributed in the last decade to highlight the physiological and pathological relevance of LLPS, which was found to occur in the cell cytoplasm, in the nucleoplasm [20][88], as well as in vitro for many purified proteins [21][114]. The growing interest towards LLPS has fueled studies aimed at gathering information on proteins undergoing LLPS in a structured and knowledgeable manner, while providing a wide range of information on the biophysical driving forces, the biological function and the regulation of these systems. These efforts have led to the development of dedicated databases [22][115], such as PhaSePro (https://phasepro.elte.hu) [23][116] and LLPSDB (http://bio-comp.org.cn/llpsdb/) [24][117], two manually curated repositories, based on experimentally verified cases of LLPS.
In analogy with the systems explored by classical physical chemistry, cellular LLPS causes homogeneous solution of macromolecules to separate into two phases. Liquid droplets form, which are enriched in condensed biomolecules and surrounded by a “diluted” phase, depleted of those components [25][1][3][93,99,101]. As long as a genuine liquid nature is conserved, polymer molecules can diffuse within the dense phase and between the dense and diluted phases [3][101]. It is mesoscopically evident that the de-mixing occurs rapidly and dynamically, in response to concentration changes of some cellular components, or to environmental stimuli, which ultimately result in changes of molecular interaction affinity. Fluid droplets can form/dissolve reversibly as they are held together by a network of weak and transient interactions [5][26][27][28][103,118,119,120]. Several in-vivo and in-vitro studies suggest that LLPS preferentially involves IDPs/IDRs because of their peculiar conformational properties [29][21][26][30][31][32][33][84,114,118,121,122,123,124]. Structural studies on the Tau protein suggest that phase separation is driven by its conformational expansion, coupled with solvation and large-scale fluctuations, while transient, non-covalent intermolecular contacts maintain the internal fluidity [34][125]. Nonetheless, LLPS has also been observed for ordered, globular proteins. Bovine serum albumin was used as a model to experimentally describe the kinetics of temperature-driven de-mixing [35][126]. Overall, condensate immiscibility appears as a very general feature in biological systems, which could also be exploited to create synthetic membrane-less compartments [36][37][127,128]. Theoretical and computational studies on protein de-mixing [36][38][39][40] [127,129,130,131] confirm that protein phase diagrams are mainly shaped by salt concentration and solvent-mediated interactions, in turn influenced by temperature, their effects being cumulatively translated into “interfacial tension”, i.e., the work required to increase the size of the interface between two adjacent, immiscible phases. [36][127]. Sequence-specific properties, such as the presence of charged and aromatic residues, may then stabilize the de-mixing arrangements [39][130]. The role of salts on protein de-mixing has been considered for the peculiar case of multivalent ions [132,133]. It has emerged that some multivalent cations (e.g., yttrium) bind to proteins by an entropically-driven process, due to changes in the structure of the hydration shell. Once the protein–cation complex is formed, the multivalent ions can act as bridging elements to drive intermolecular interactions [41][42][132,133].
Networking is a very relevant property in LLPS, mainly related to the modularity and the “multivalence” of the involved macromolecules. Multivalence refers to the presence of multiple, repetitive interaction sites within a polypeptide or nucleic acid [43][44][134,135]. Again, IDPs and IDRs, with their low-complexity domains (LCDs) and multiple interacting sites (i.e., short linear motifs, SLiMs) [45][46][47][136,137,138], have a privileged role among cellular components undergoing LLPS [43][45][134,136], causing the formation of densely packed liquid phases [48] [139] often undergoing “gelation” when the network of interactions span the entire system, i.e., the whole droplet [49][140]. In this context, multivalent proteins have also been described as a peculiar type of “associative polymers” endowed with “stickers-and-spacers architecture”, where the stickers are motifs and interaction sites, and the linkers are those protein regions contributing to sticker networking [50][141].
The “molecular grammar” of interactions governing LLPS is now beginning to be understood, although it is still in its infancy [51][142]. IDPs that drive LLPS typically display low-complex sequences, characterized by long stretches with overall low diversity of amino acids [52][78]. The sequences are often repetitive and are enriched in glycine, polar sidechains (glutamine, asparagine and serine), positively charged sidechains (Arg and Lys), negatively charged sidechains (Asp and Glu), and aromatic sidechains (Phe and Tyr). The sequences of interest often encompass multiple short motifs such as YG/S-, FG-, RG-, GY-, KSPEA-, SY- and Q/N-rich regions, and blocks of alternating charges [53][143]. Interactions occur via different residue types, such as Phe and Tyr involved in π-π stacking and π-cationic interactions with Lys and Arg residues, polar and charged residues [48][51][54][55][56][139,142,144,145,146]. Distribution of charged amino acids along the sequence has also been suggested to have a role in the coacervation propensity [57][147], with electrostatic interactions between blocks of oppositely charged residues being associated to stronger driving forces—through long-range, interchain attractions [53] [143] and larger coacervation windows [38][129] than sequences with interspersed charges. This is in agreement with the general envision that phase behaviour of multivalent proteins is governed by patterning rules of “sticker” motifs of different nature [58][148]. The compositional rules emerging from in-vitro and in-cell studies also suggest the existence of two kinds of components, with distinct roles: multivalent proteins forming scaffolds and low-valence, client molecules tethered onto those scaffolds. The state of a droplet can change rapidly, through changes in the composition and concentration of scaffold proteins or through changes in their valence [59][149].
3. Techniques to Study LLPS
LLPS can be documented and monitored using various methods of varying complexity. These include simple macroscopic observation of purified samples and assessment of their turbidity, measurements of optical density and light scattering, light microscopy (fluorescence-based or contrast-based) and super-resolution microscopy studies. LLPS can be experimentally characterized both in vitro and in vivo. For in-vitro assays, the minimal components are combined in buffers to recapitulate LLPS, whereas in-vivo studies are carried out in cells, tissues or even animals. Here we provide a concise description of the most commonly used methods. A more exhaustive description of the available methods can be found in the excellent reviews by Alberti et al. and Mitrea et al. [10][60][108,150].
For in-vitro studies, a very simple clue of LLPS is the appearance of sample turbidity. Phase separated samples become turbid because the droplets (i.e., the coacervates) scatter light. They can be detected by optical density measurements (typically at wavelengths of 340, 400 or 600 nm) or by direct static light scattering. The latter approach allows determination of the onset of scattering of a dilution series, thus enabling identification of the threshold concentration for droplet formation, Csat.
One caveat of simple turbidity measurements resides in the fact that they detect a variety of assemblies and do not differentiate their shape, size or the mechanism underlying their formation. Turbidity measurements should thus be used in conjunction with microscopy. Under a microscope, the separation process is materialized by the appearance of randomly moving, spherical and dynamic droplets, of micrometric size. Coacervates are to be distinguished from aggregates that are often irreversible and irregularly shaped. Quantitative image analysis enables assessing coacervate sphericity, deformability under shear stress and coalescence abilities, three properties commonly regarded as reflecting the liquid nature of the bio-condensates. One caveat is that it may be impossible to detect diffraction-limited assemblies with simple methods, and super-resolution microscopy, which combines optical inputs with mathematical analysis to construct images of specimens with resolution from 2- to 10-fold below the diffraction limit, may be required.
An easy way to demonstrate in vitro that an assembly forms through LLPS is to define a saturation concentration Csat, where for C < Csat the protein is diffuse in solution, and for C > Csat dense droplets can be observed. The volume fraction of these dense droplets increases when the concentration is increased further, while the concentration in the light phase (CL) remains constant. This behavior is typical of phase separation and constitutes a strong support for LLPS. The increase in the volume fraction of the dense phase (CD) can be estimated via light scattering or centrifugation. The use of fluorescently labeled components allows estimation of the concentration inside CD, provided that standard curves for the fluorophore at different concentrations are obtained under identical imaging conditions.
Centrifugation of systems in the two-phase regime, and the ensuing separation of light and dense phases, allows precise measurements of the concentrations of two coexisting phases, CL and CD, as a function of a given physico-chemical parameter. LLPS of the stock solution is induced by changing the solution conditions, and after incubation, the dense phase is sedimented by centrifugation [61][151]. An aliquot of the light phase is removed, and its concentration determined spectroscopically (e.g., by measuring the absorbance at 280 nm or fluorescence intensity if dealing with a fluorophore-labeled protein). This experimental design enables assessing whether CL remains constant when using different protein concentrations [32][62][123,152]. It should be emphasized that processes such as fibrillation may interfere with LLPS, making accurate determination of CL difficult. Thus, microscopic analysis that can distinguish liquid droplets from aggregates or fibers is, once again, recommended.
Fluorescence Recovery After Photobleaching (FRAP) is commonly regarded as a technique for ascertaining the liquid versus solid nature of biomolecular condensates. FRAP monitors the diffusion of fluorescent molecules within a photobleached region and is used to assess macromolecular mobility within phase-separated condensates both in vitro and in vivo. A specific region of interest is illuminated by a high-intensity laser, at the excitation wavelength of the fluorophore, to irreversibly convert the molecules to a dark state. The diffusion of fluorescent molecules from outside to inside this region is then quantified by measuring the variation in fluorescence intensity as a function of time. From the evolution of the fluorescence intensity within the region of interest, the rate of (or half-time for) fluorescence recovery of a photobleached component, and the extent of fluorescence recovery (referred to as the mobile fraction), can be derived. Fast exchange rates (i.e., in the second range) characterize liquid-like assemblies. If the droplet is large enough, bleaching within droplets allows probing intra-droplet dynamics [16][63][110,153]. Importantly, FRAP can also be useful in assessing whether the droplets are spatially homogeneous based on the pattern of recovery.
Many proteins, either in the presence or absence of RNA, are capable of undergoing LLPS in vitro when a sufficiently high concentration is reached. Therefore, the mere observation that a given protein undergoes LLPS in vitro does not imply per se a functional relevance, and proving LLPS occurrence in vivo is, therefore, of paramount importance. Demonstrating that an assembly results from LLPS in the cellular context is challenging and much caution should be taken before drawing definite conclusions, especially when interpreting over-expression data. An ideal approach would be to attempt at establishing phase diagrams in living cells [27][119]. To this end, it is necessary to vary in a controlled manner the expression levels of the protein of interest. The measured Csat can then be directly compared to the cellular concentration of the protein under a given condition, enabling to conclude whether phase separation can take place or not.
Currently, the commonly accepted criteria for considering that an assembly is a phase-separated structure are (i) spherical shape, (ii) ability to fuse (coalescence), (iii) deformability under shear stress and (iv) fast recovery from photobleaching. However, it should be kept in mind that fast recovery rates from FRAP can arise from multiple reasons and several potential sources of artifacts may affect the quantitative interpretation of photobleaching results (reviewed in [10][60][108,150]). Thus, none of the above features is sufficient on its own to unequivocally demonstrate that a structure is formed via LLPS.
4. Functional and Dysfunctional Aspects of LLPS
The structural and material properties of LLPS condensates resulting from LLPS are intimately linked to their function as reaction crucible and organizational hub [1][99]. For instance, signaling complexes trapped inside condensates can better coordinate cell response to environmental stimuli [64][154]. Overall, sequestration inside LLPS droplets, because of the high viscosity, can exclude some interactions (with “external” molecules) and increase some others, by enhancing the probability and kinetics of molecular interactions and enzymatic reactions among “internal” molecules [65][66][67][155,156,157].
The “maturation” of liquid droplets toward gel or solid state has also been reported [68][92,102,108,140,158]. Amyloid-like fibers can form hydrogels, which are non-dynamic and rather stable, as they dissolve only under harsh conditions (i.e., high salt or denaturant) [69][70][71][159,160,161]. Liquid-to-solid transitions have been proven functional or dysfunctional/pathological for several different proteins. One example of functional liquid-to-solid transitions is offered by SGs, formed upon heat stress by the yeast protein Pab1. This protein forms glassy droplets that play an adaptative role, promoting cell survival upon heat shock [72][108,162]. Dysfunctionally persistent SGs were obtained from the RNA-binding protein hnRNPA1, whose fibrillation is enhanced in protein-rich droplets [121]. Mutations in fused-in-sarcoma RNA-binding protein (FUS) and in heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) are involved in familial forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) [73][74][75][163,164,165]. Both proteins form liquid droplets at moderate concentrations, but these droplets can nucleate amyloid-like fibers and begin to jellify over time, a process accelerated when the protein exhibits disease-associated mutations [76][119,121,166]. IDRs involved in LLPS are therefore implicated in neurodegeneration and able to undergo fibrillation [77][119,121,152,160,167]. Consistently, many protein domains that promote phase separation have been described as prion-like [78][168].
The liquid-droplet environment also strongly accelerates fibrillation of TAR DNA-binding protein of 43 kDa (TDP-43). Rapid assembly of fibrillar aggregates involves the low complexity domain of TDP-43 and develops from mature liquid droplets [79][169]. Overall, one can assume that the occurrence of LLPS is prodromal to a wide range of possible transitions, giving rise to a continuum of material properties, according to the nature of involved proteins and the mechanisms of their interactions [80][99,170].
The rapid assembly and disassembly of liquid condensates compared to membrane-delimited organelles enables a rapid cellular reorganization and hence a rapid response to stimuli. Because IDPs/IDRs are known to be heavily targeted by PTMs and alternative splicing, and because both kinds of events can rewire interactomes, the involvement of IDPs/IDRs in LLPS offers an exquisite manner to regulate the spatio-temporal formation of these cellular condensates [81][154,171].
Considering that viral proteins are enriched in IDRs and are multivalent, their ability to undergo LLPS is not surprising, as well illustrated by recent studies that have provided experimental evidence for this phenomenon and have elaborated on its functional implications.