RNA modifications are essential for proper RNA processing, quality control, and maturation steps. In the last decade, some eukaryotic DNA repair enzymes have been shown to have an ability to recognize and process modified RNA substrates and thereby contribute to RNA surveillance. Single-strand-selective monofunctional uracil-DNA glycosylase 1 (SMUG1) is a base excision repair enzyme that not only recognizes and removes uracil and oxidized pyrimidines from DNA but is also able to process modified RNA substrates. SMUG1 interacts with the pseudouridine synthase dyskerin (DKC1), an enzyme essential for the correct assembly of small nucleolar ribonucleoproteins (snRNPs) and ribosomal RNA (rRNA) processing. Here, we review rRNA modifications and RNA quality control mechanisms in general and discuss the specific function of SMUG1 in rRNA metabolism. Cells lacking SMUG1 have elevated levels of immature rRNA molecules and accumulation of 5-hydroxymethyluridine (5hmU) in mature rRNA. SMUG1 may be required for post-transcriptional regulation and quality control of rRNAs, partly by regulating rRNA and stability.
- SMUG1
- rRNA processing
- modified bases
1. Introduction
A wide variety of functional base modifications are present in cellular RNA in addition to the regular four ribonucleosides. Over 160 known chemical modifications that modulate the structure and function of RNA molecules have been described [1][2][3][4][5][6][7]. Although most of the modifications described so far are found in abundant non-coding RNAs (ncRNAs), such as transfer (tRNAs) and ribosomal RNAs (rRNAs), recent advances in enrichment/capture techniques coupled with next-generation sequencing strategies have revealed an increasing number of different modifications both on coding and non-coding RNAs. Thus, all RNA classes, including messenger (mRNAs) and small nuclear RNAs, contain base modifications. For example, N6-methyladenosine (m6A), N1-methyladenosine (m1A) [8], 5-methylcytidine (m5C) [9][10][11], 5-hydroxylmethylcytidine (hm5C) [12], and inosine [13] are found in mRNA [14]. Base modifications introduced enzymatically at defined positions change RNA function at several levels. Here, we will first give an overview of the main rRNA modifications and RNA quality control mechanisms and then discuss recent developments implicating the SMUG1 DNA-glycosylase in rRNA biogenesis. In SMUG1 knock-down cells, immature and mature rRNAs accumulated 5-hydroxylmethyluridine (hm5U), a base modification recognized by SMUG1, pointing to SMUG1 as a possible new enzyme involved in the regulation of rRNA.
A wide variety of functional base modifications are present in cellular RNA in addition to the regular four ribonucleosides. Over 160 known chemical modifications that modulate the structure and function of RNA molecules have been described [1,2,3,4,5,6,7]. Although most of the modifications described so far are found in abundant non-coding RNAs (ncRNAs), such as transfer (tRNAs) and ribosomal RNAs (rRNAs), recent advances in enrichment/capture techniques coupled with next-generation sequencing strategies have revealed an increasing number of different modifications both on coding and non-coding RNAs. Thus, all RNA classes, including messenger (mRNAs) and small nuclear RNAs, contain base modifications. For example, N6-methyladenosine (m6A), N1-methyladenosine (m1A) [8], 5-methylcytidine (m5C) [9,10,11], 5-hydroxylmethylcytidine (hm5C) [12], and inosine [13] are found in mRNA [14]. Base modifications introduced enzymatically at defined positions change RNA function at several levels. Here, we will first give an overview of the main rRNA modifications and RNA quality control mechanisms and then discuss recent developments implicating the SMUG1 DNA-glycosylase in rRNA biogenesis. In SMUG1 knock-down cells, immature and mature rRNAs accumulated 5-hydroxylmethyluridine (hm5U), a base modification recognized by SMUG1, pointing to SMUG1 as a possible new enzyme involved in the regulation of rRNA.
2. Ribosomal RNA Modifications and Their Biological Functions
In eukaryotic rRNA, 2% of all rRNA nucleotides are modified [6]. Several modifications have been mapped: These include enzymatically deposited base modification, exemplified by pseudouridines (Ψ), spontaneously introduced base modifications, e.g., oxidized bases, and residues methylated at the ribose phosphate backbone, such as 2′-
O
-methylation of ribose moieties (Am, Gm, Cm, and Um) (
A). Deposition of RNA modifications is regulated at several levels. In human cells, the majority of the small nucleolar RNA complex (snoRNP)-guided modifications, e.g., pseudouridylation and 2′-
O-methylation, occur at early ribogenesis steps [6]. Only a few snoRNAs have been shown to target late pre-ribosomal intermediates where the activity of other factors, such as the RNA helicases, is required for structural rearrangements of the target to allow the snoRNAs to access the substrate [6][15]. On the other hand, other base modifications occur at later stages of ribosomal maturation, although the specific stage and timing has not been elucidated [6][16].
-methylation, occur at early ribogenesis steps [6]. Only a few snoRNAs have been shown to target late pre-ribosomal intermediates where the activity of other factors, such as the RNA helicases, is required for structural rearrangements of the target to allow the snoRNAs to access the substrate [6,15]. On the other hand, other base modifications occur at later stages of ribosomal maturation, although the specific stage and timing has not been elucidated [6,16].
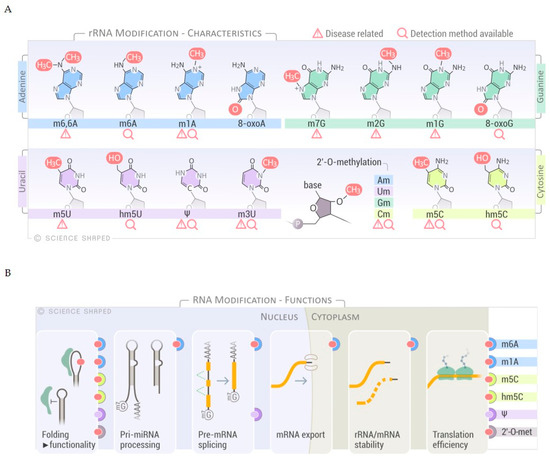
Figure 1.
Overview of RNA modifications, substrates and functions. (
A
) Selected spectrum of RNA chemical modifications found in ribosomal RNA (rRNA). Disease relationships and sequencing methods are shown. (
B
) Schematic representation of the different functions of the selected RNA modifications. The modifications known to have a role in specific RNA processes are shown on the right-hand side of the panel.
Further regulation of RNA modification is achieved through a specific association of the target RNA molecule and guide snoRNA in determined cellular locations, a molecular matchmaking that has been extensively described for pseudouridylation [17][18][19]. Modifications on rRNA are mainly concentrated in functional regions of the ribosome such as the peptidyl transferase center, the intersubunit interface, and the decoding and tRNA binding sites [6][20][21]. Modifications regulate not only the efficiency and accuracy of translation, as a consequence of ribosomal structure and function, but also rRNA processing and cleavage [6][22]. These modifications alter the structure/conformation and stability of the rRNAs due to changes in the molecular interactions within functional domains and distant regions of the ribosome. In addition, rRNA modifications may change the affinity of ribosomes to specific mRNA structures (i.e., internal ribosome entry sites), thereby governing the protein synthesis of a particular subset of mRNAs [6][23]. Interestingly, a role for rRNA modifications in ribosome heterogeneity by altering the ribosomal activity in response to environmental stressors has recently emerged, expanding the functions of modified bases in rRNA regulation to include fine-tuning of the translation cycle and modulating gene expression in response to external cues (
Further regulation of RNA modification is achieved through a specific association of the target RNA molecule and guide snoRNA in determined cellular locations, a molecular matchmaking that has been extensively described for pseudouridylation [17,18,19]. Modifications on rRNA are mainly concentrated in functional regions of the ribosome such as the peptidyl transferase center, the intersubunit interface, and the decoding and tRNA binding sites [6,20,21]. Modifications regulate not only the efficiency and accuracy of translation, as a consequence of ribosomal structure and function, but also rRNA processing and cleavage [6,22]. These modifications alter the structure/conformation and stability of the rRNAs due to changes in the molecular interactions within functional domains and distant regions of the ribosome. In addition, rRNA modifications may change the affinity of ribosomes to specific mRNA structures (i.e., internal ribosome entry sites), thereby governing the protein synthesis of a particular subset of mRNAs [6,23]. Interestingly, a role for rRNA modifications in ribosome heterogeneity by altering the ribosomal activity in response to environmental stressors has recently emerged, expanding the functions of modified bases in rRNA regulation to include fine-tuning of the translation cycle and modulating gene expression in response to external cues (
B) [6].
Several reports connect defects in the rRNA modification machinery, which encompasses snoRNAs and protein components of the snoRNP complexes or stand-alone rRNA-modifying enzymes, with genetic diseases and cancers. However, it is still unclear whether pathogenic effects are driven by the lack of modification per se [6].
Here, a selected set of known rRNA modifications and their functions are presented (for a comprehensive list of all the rRNA modifications and the sequencing methods used for their detection, the reader is redirected to [24]).
2.1. Pseudouridylation
Pseudouridine is considered the fifth ribonucleoside due to its abundance, and it is found in both the large and small subunits of the ribosome [6][19][25][26]. The role of Ψ in rRNA is still under debate, but it is classically described to improve base stacking due to increased backbone rigidity and to stabilize the secondary and tertiary structures for ribosomal subunit association [22][25][27].
Pseudouridine is considered the fifth ribonucleoside due to its abundance, and it is found in both the large and small subunits of the ribosome [6,19,25,26]. The role of Ψ in rRNA is still under debate, but it is classically described to improve base stacking due to increased backbone rigidity and to stabilize the secondary and tertiary structures for ribosomal subunit association [22,25,27].
Pseudouridine is enzymatically introduced by the isomerization of uridine. In humans, the main pseudouridylase is dyskerin (DKC1) [28]. DKC1 exerts its function as part of a ribonucleoprotein (RNP) complex, which consists of four core proteins (GAR1, NHP2, and NOP10 in addition to DKC1) and a short RNA molecule (H/ACA snoRNA). The H/ACA snoRNA contains a conserved 5′-ANANNA-3′ sequence called “hinge box” (or H-box) and a sequence of three nucleotides (ACA) present at its 3′-end (called ACA-box). This snoRNA functions as a guide RNA that defines the residue to be modified through base pairing in the “pseudouridylation pocket,” while the protein complex ensures the correct positioning of the target nucleotide [6][27][29]. Even though most of the pseudouridines in rRNA are modified by the H/ACA-snoRNA-guided machinery, a contribution from stand-alone human pseudouridine synthases (i.e., PUS1 and PUS7) cannot be completely excluded [6][25]. How specificity is achieved for PUS enzymes remain to be discovered [30].
Pseudouridine is enzymatically introduced by the isomerization of uridine. In humans, the main pseudouridylase is dyskerin (DKC1) [28]. DKC1 exerts its function as part of a ribonucleoprotein (RNP) complex, which consists of four core proteins (GAR1, NHP2, and NOP10 in addition to DKC1) and a short RNA molecule (H/ACA snoRNA). The H/ACA snoRNA contains a conserved 5′-ANANNA-3′ sequence called “hinge box” (or H-box) and a sequence of three nucleotides (ACA) present at its 3′-end (called ACA-box). This snoRNA functions as a guide RNA that defines the residue to be modified through base pairing in the “pseudouridylation pocket,” while the protein complex ensures the correct positioning of the target nucleotide [6,27,29]. Even though most of the pseudouridines in rRNA are modified by the H/ACA-snoRNA-guided machinery, a contribution from stand-alone human pseudouridine synthases (i.e., PUS1 and PUS7) cannot be completely excluded [6,25]. How specificity is achieved for PUS enzymes remain to be discovered [30].
Pseudouridine-related enzymes are implicated in human diseases. X-linked dyskeratosis congenita (X-DC), a severe disorder characterized by bone marrow failure, lung fibrosis, and increased susceptibility to cancer, is caused by mutations in
DKC1. Patients present lower levels of Ψ compared to the healthy controls that may ultimately impair internal ribosome entry site (IRES)-mediated translation of a subset of mRNAs, such as TP53 and CDKN1B [6][18][20][23][31][32][33][34]. Illustrating the complex pathogenicity of pathways involving RNA modification enzymes, many symptoms of X-DC patients are related to the function of DKC1 in telomere maintenance and not to the deposition of pseudouridines in rRNA per se [22][35][36][37]. Two PUS enzymes are associated with human diseases;
. Patients present lower levels of Ψ compared to the healthy controls that may ultimately impair internal ribosome entry site (IRES)-mediated translation of a subset of mRNAs, such as TP53 and CDKN1B [6,18,20,23,31,32,33,34]. Illustrating the complex pathogenicity of pathways involving RNA modification enzymes, many symptoms of X-DC patients are related to the function of DKC1 in telomere maintenance and not to the deposition of pseudouridines in rRNA per se [22,35,36,37]. Two PUS enzymes are associated with human diseases;
PUS1
and
PUS3 mutations are found in patients with the mitochondrial disease MLASA (Mitochondrial myopathy, lactic acidosis and sideroblastic anemia) [38] and with intellectual disability [39], respectively.
mutations are found in patients with the mitochondrial disease MLASA (Mitochondrial myopathy, lactic acidosis and sideroblastic anemia) [38] and with intellectual disability [39], respectively.
While early work was restricted to studies of pseudouridine in highly abundant long-lived RNA species due to limited sensitivity and specificity of the methods, recent technological developments in the targeted sequencing of RNA modifications have allowed the identification of pseudouridylated modifications at single-nucleotide resolution present in sub-stoichiometric amounts in non-coding RNA as rRNA, tRNA, and small nuclear RNA. The most common sequencing technique for the detection of pseudouridines is based on the derivatization of Ψ with carbodiimide and mutation insertion or block of reverse transcription during high-throughput sequencing [18][40][41][42]. Different efficiencies in carbodiimide incorporation makes this technique semi-quantitative. To alleviate this limitation, a novel method based on hydrazine/aniline cleavage was recently developed for systematic mapping and absolute quantification of Ψ, where the signals obtained by negative hits correspond directly to Ψ residues, protected from the hydrazine-dependent cleavage [43].
While early work was restricted to studies of pseudouridine in highly abundant long-lived RNA species due to limited sensitivity and specificity of the methods, recent technological developments in the targeted sequencing of RNA modifications have allowed the identification of pseudouridylated modifications at single-nucleotide resolution present in sub-stoichiometric amounts in non-coding RNA as rRNA, tRNA, and small nuclear RNA. The most common sequencing technique for the detection of pseudouridines is based on the derivatization of Ψ with carbodiimide and mutation insertion or block of reverse transcription during high-throughput sequencing [18,40,41,42]. Different efficiencies in carbodiimide incorporation makes this technique semi-quantitative. To alleviate this limitation, a novel method based on hydrazine/aniline cleavage was recently developed for systematic mapping and absolute quantification of Ψ, where the signals obtained by negative hits correspond directly to Ψ residues, protected from the hydrazine-dependent cleavage [43].
2.2. 2′-O-Methylation
Ribose 2′-
O
-methylation (2′-
O
-Me) at any nucleotide (Am, Gm, Um, and Cm), is another highly abundant modification with more than 100 sites reported for human rRNA (
O
-Me might be involved in stabilizing the secondary and tertiary structures of rRNA, essential for ribosomal function (
B). As demonstrated on synthetic substrates, 2′-
O-Me impaired the stability and flexibility of the stem-loops by preventing hydrolysis of the phosphate backbone and favoring an endo conformation at the 3′-end [22][45]. These methylations are formed by either stand-alone enzymes or by C/D-box snoRNP complexes that base-pair with the pre-rRNA and re-direct the RNA modifying enzyme to the specific target residue, the same principle as described for pseudouridylation guided by H/ACA box snoRNP [44]. The C/D box RNA has a bipartite structure containing a C-box (5′-RUGAUGA-3′, where R is a purine), a D-box (5′-CUGA-3′) at both ends, and related C′- and D′-boxes in the internal regions. The spacer regions between the boxes contain a guide sequence that can range from 10 to 21 nucleotides [46]. The proteins that form the RNP complex (NOP56, NOP58, and 15.5K) facilitate base-pairing and positioning of the catalytic site of the methyltransferase fibrillarin (FBL) to its target [6]. Although only 10 nucleotides form guide–substrate duplexes, the extensive base-pairing enhances the specificity of target recognition and prevents misfolding of the rRNA by sequestering the target [6][46]. Interestingly, a recent study in
-Me impaired the stability and flexibility of the stem-loops by preventing hydrolysis of the phosphate backbone and favoring an endo conformation at the 3′-end [22,45]. These methylations are formed by either stand-alone enzymes or by C/D-box snoRNP complexes that base-pair with the pre-rRNA and re-direct the RNA modifying enzyme to the specific target residue, the same principle as described for pseudouridylation guided by H/ACA box snoRNP [44]. The C/D box RNA has a bipartite structure containing a C-box (5′-RUGAUGA-3′, where R is a purine), a D-box (5′-CUGA-3′) at both ends, and related C′- and D′-boxes in the internal regions. The spacer regions between the boxes contain a guide sequence that can range from 10 to 21 nucleotides [46]. The proteins that form the RNP complex (NOP56, NOP58, and 15.5K) facilitate base-pairing and positioning of the catalytic site of the methyltransferase fibrillarin (FBL) to its target [6]. Although only 10 nucleotides form guide–substrate duplexes, the extensive base-pairing enhances the specificity of target recognition and prevents misfolding of the rRNA by sequestering the target [6,46]. Interestingly, a recent study in
Saccharomyces cerevisiae indicates that a subset of snoRNAs can use a single guide to induce multiple modifications in the target region by forming two different snoRNP complexes that differ with respect to the positioning of the protein components (NOP56 and FBL). This mechanism may also be possible in other eukaryotes, which could increase the complexity of rRNA modifications without the requirement of additional snoRNAs [47].
indicates that a subset of snoRNAs can use a single guide to induce multiple modifications in the target region by forming two different snoRNP complexes that differ with respect to the positioning of the protein components (NOP56 and FBL). This mechanism may also be possible in other eukaryotes, which could increase the complexity of rRNA modifications without the requirement of additional snoRNAs [47].
The development of new high-throughput approaches has substituted laborious methods based on RNase H cleavage and retrotranscription. Through detection and systematic mapping of 2′-
O-Me in different samples, it was shown that hypomodified regions lie peripherally on the 3-D structure of the ribosomes while the functional centers are heavily modified [48][49][50][51][52][53][54]. These methods confirmed the co-existence of distinct subsets of ribosomes that are only partially modified and may potentially exert specific functions [23][50][54]. Changes in 2′-
-Me in different samples, it was shown that hypomodified regions lie peripherally on the 3-D structure of the ribosomes while the functional centers are heavily modified [48,49,50,51,52,53,54]. These methods confirmed the co-existence of distinct subsets of ribosomes that are only partially modified and may potentially exert specific functions [23,50,54]. Changes in 2′-
O-Me profiles in rRNA have been linked to ribosomopathies such as Treacher Collins syndrome and cancer susceptibility [6][55][56].
-Me profiles in rRNA have been linked to ribosomopathies such as Treacher Collins syndrome and cancer susceptibility [6,55,56].
3. rRNA Processing and Maturation
Ribosome biogenesis and assembly of the small and large subunits of the eukaryotic ribosome takes place in the nucleus before its final maturation in the cytoplasm. It requires the association of 80 ribosomal proteins (RPs) with four distinct ribosomal RNAs. The small subunit (40S, SSU) is formed by the association of the 18S rRNA with 33 RPs; the large subunit (60S, LSU) contains the 5S, 5.8S, and 25S/28S rRNAs associated with 47 RPs [57]. The ribosomal genes are arranged as direct head-to-tail tandem ribosomal DNA (rDNA) repeats at the nucleolar organizer regions (NORs), and they are present in several copies within eukaryotic genomes (>200 rRNA genes/genome of five distinct chromosomes). Only a fraction of these genes is actively transcribed [58]. rRNA biogenesis starts in the nucleolus where RNA polymerase I (RNAPI or PolI) transcribes a long primary transcript that has to be processed in order to produce the mature rRNAs (18S, 5.8S, and 25S/28S rRNAs) (
Ribosome biogenesis and assembly of the small and large subunits of the eukaryotic ribosome takes place in the nucleus before its final maturation in the cytoplasm. It requires the association of 80 ribosomal proteins (RPs) with four distinct ribosomal RNAs. The small subunit (40S, SSU) is formed by the association of the 18S rRNA with 33 RPs; the large subunit (60S, LSU) contains the 5S, 5.8S, and 25S/28S rRNAs associated with 47 RPs [96]. The ribosomal genes are arranged as direct head-to-tail tandem ribosomal DNA (rDNA) repeats at the nucleolar organizer regions (NORs), and they are present in several copies within eukaryotic genomes (>200 rRNA genes/genome of five distinct chromosomes). Only a fraction of these genes is actively transcribed [97]. rRNA biogenesis starts in the nucleolus where RNA polymerase I (RNAPI or PolI) transcribes a long primary transcript that has to be processed in order to produce the mature rRNAs (18S, 5.8S, and 25S/28S rRNAs) (
). The primary transcript (47S) comprises the mature rRNAs separated by the internal transcribed spacer 1 (ITS1) and 2 (ITS2) flanked by the 5′- and 3′-external transcribed spacers (5′- and 3′-ETS) (
). The primary transcript is sequentially cleaved via a complex sequence of endonucleolytic and exonucleolytic cleavages (
Figure 2) [57][59][60][61]. The 47S rRNA is cleaved at the 5′- and 3′-ends (sites 01 and 02, respectively) to form the 45S precursor that can be processed via two main pathways (pathway 1 and 2), depending on the cleavage sites used. In pathway 1, processing starts at the 5′-end of the molecule with the cleavage at sites A0 and 1, forming 41S rRNA followed by successive trimming at site 2 into 21S and 32S pre-rRNAs. In pathway 2, the cleavage begins at site 2, located within the ITS1. The newly generated 30S pre-rRNA is further processed directly to 21S pre-rRNA through cutting at the A0 and 1 sites, or via an intermediate form called 26S pre-rRNA, where the cleavage at these sites is uncoupled (
) [96,98,99,100]. The 47S rRNA is cleaved at the 5′- and 3′-ends (sites 01 and 02, respectively) to form the 45S precursor that can be processed via two main pathways (pathway 1 and 2), depending on the cleavage sites used. In pathway 1, processing starts at the 5′-end of the molecule with the cleavage at sites A0 and 1, forming 41S rRNA followed by successive trimming at site 2 into 21S and 32S pre-rRNAs. In pathway 2, the cleavage begins at site 2, located within the ITS1. The newly generated 30S pre-rRNA is further processed directly to 21S pre-rRNA through cutting at the A0 and 1 sites, or via an intermediate form called 26S pre-rRNA, where the cleavage at these sites is uncoupled (
Figure 2) [57][59][60][61]. Intriguingly, a role for the exosome and DIS3L2/ERI1 in the 5.8S maturation steps via the formation of a cytoplasmic precursor, named 7SB, was recently demonstrated [62].
) [96,98,99,100]. Intriguingly, a role for the exosome and DIS3L2/ERI1 in the 5.8S maturation steps via the formation of a cytoplasmic precursor, named 7SB, was recently demonstrated [101].
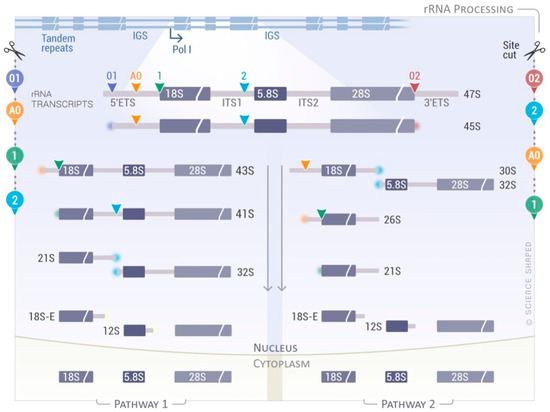
Figure 2.
Simplified rRNA processing pathway in eukaryotes. RNA Polymerase I (Pol I) transcribes a long primary transcript (47S pre-rRNA) from loci containing hundreds of rDNA copies. This transcript contains the sequences for the mature 18S, 5.8S, and 28S rRNAs flanked by external (5′- and 3′-ETS) and internal spacers (ITS1 and ITS2), which are enzymatically removed by endo- and exonucleases. Depending on the cleavage sites used and the kinetics, two main pathways can be depicted forming different precursors. Cleavage of the 45S rRNA precursor starts either in the 5′-ETS at cleavage site 1 (Pathway 1, left side) or in the ITS1 sequence at cleavage site 2 (Pathway 2, right side). The cleavage sites used for each pathway are shown on the side.
For an exhaustive description of the rRNA processing pathways, the reader is redirected to recent review articles [57][59][60][61][62].
For an exhaustive description of the rRNA processing pathways, the reader is redirected to recent review articles [96,98,99,100,101].