Interferons (IFN) are crucial for the innate immune response. Slightly more than two decades ago, a new type of IFN was discovered: the lambda IFN (type III IFN). Like other IFN, the type III IFN display antiviral activity against a wide variety of infections, they induce expression of antiviral, interferon-stimulated genes (MX1, OAS, IFITM1), and they have immuno-modulatory activities that shape adaptive immune responses. Unlike other IFN, the type III IFN signal through distinct receptors is limited to a few cell types, primarily mucosal epithelial cells. Type III IFN elicit fewer inflammatory responses than the Type I IFN and are gaining interest as antiviral treatments.
- respiratory viruses
- influenza
- respiratory viruses,influenza
1. Introduction
1.1. Virus Entry Triggers Host Signaling Responses
In viral infection, the protective barriers are host skin and mucous membranes. In the initial stages of a viral infection, quick activation of a non-specific immune response occurs in response to the infiltration in a cell (Figure 1). Cells utilize various pattern recognition receptors (PRR) to detect viral particles. Toll-like receptors (TLR) are one such sensor. RIG-I-like receptors (RLR), Nod-like receptors, and cytosolic nucleic acid sensors become involved when viral particles enter the cytoplasm [1]. To date, ten TLR types have been found in humans. It is well known that TLR-3, TLR-7, TLR-8, and TLR-9 are localized in endosomes, while others reside on the outer surface of the cytoplasmic membrane [1][2][1,2]. Signaling through TLR requires the use of various combinations of the following protein adapters: MyD88, Mal, TRIF, and TRAM [2]. Activation of molecular adapters is aimed at regulating the activity of the NFκB, IRF-3, and IRF-7 transcription factors, and activation of MAPK-dependent signaling pathways. The combined action of these transcription factors with the AP-1 protein effectively induces the expression of target genes [1][3][1,3].
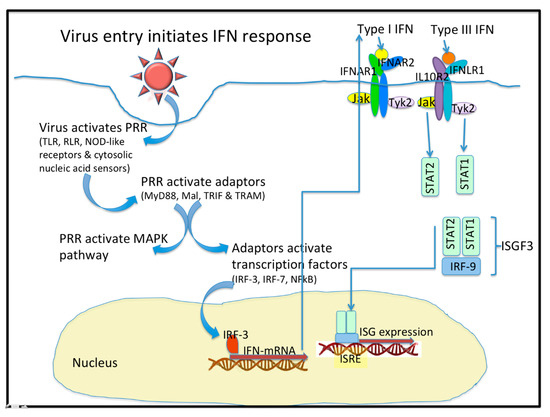
Figure 1. Virus entry initiates the interferon (IFN) response. Upon entering a cell, virus activates Pattern Recognition Receptors (PRR) that include the Toll-like receptors (TLR), the RIG-I-like receptors (RLR), the NOD-like receptors, and some cytosolic nucleic acid receptors. These PRR activate adaptor molecules like MyD88, Mal, TRIF, and TRAM. PRR also activate the MAPK pathway. The Adaptor molecules activate Transcription factors like IRF-3, IRF-7 and NF-kB. IFN mRNA are transcribed and they express the type I, type II (not shown), and type III IFN. Type I and Type III IFN bind to distinct receptors that activate similar signaling pathways and transcriptional responses. The heterodimeric IFN receptors signal through the JAK/STAT pathways to form a complex with IRF-9 to initiate the expression of hundreds of Interferon stimulated genes (ISG). The ISG are peptidic antivirals that interfere with virus replication.
Cells are capable of TLR-independent responses to pathogen infiltration, and such responses are mediated through cytosolic sensors. The most important sensors are RNA helicases that belong to the RLR family: RIG-I, MDA-5 and LGP-2 [1]. The activated multimeric forms of RIG-I or MDA-5 are able to interact with the MAVS protein adapter located on the outer mitochondrial membrane or in peroxisomes [1]. Viral dsRNA activates both RIG-I and MDA-5. RNA containing a 5′-triphosphate end, without a cap structure, can also activate RIG-I. The MAVS protein adapter plays the role of a scaffold protein and is involved in the recruitment of signaling cascade components aimed at activating both NFκB and IRF-3 [3].
1.2. IFN Are Class II Cytokines
Class II cytokines are an extensive family of protein mediators that have similar gene structure, receptor structure and common signaling pathways. Four types of cytokines are commonly assigned to this family: “IL-10-like” cytokines; and the type I, II, and III interferons (IFN) [4]. Class II cytokine receptors are heterodimers and consist of a subunit featuring high ligand affinity (usually referred to as R1) and a low-affinity subunit (R2). Both subunits, however, are necessary for signal transmission [4]. IFN play a crucial role in the immune response. They inhibit the spread of viral infection in the early stages of illness and form the first line of defense in mammals against viral infections [5][6][5,6]. All IFN have an α-helical structure. According to the amino acid sequence and the type of receptor through which signal transmission is mediated, IFN are divided into three groups [3][6][7][3,6,7].
The most studied are type I IFN. In humans, a number of genes have been identified: 13 genes encoding different IFN-α subtypes; 1 gene encoding IFN-β; and other genes encoding IFN-ω, IFN-τ, IFN-ε, IFN-δ and IFN-κ [8]. Despite the wide variety of type I IFN, their action is mediated through the ubiquitous, heterodimeric IFNα receptor (IFNαR); such action is aimed at the induction of interferon-stimulated genes (ISG) [6]. The biological activity of type I IFN depends on their affinity for receptor subunits and the density of these subunits on the cell surface [9]. For example, all IFN-α subtypes are characterized by a non-optimal affinity for the subunit of the receptor [4] [4], which leads to differences in biological activity between α and β IFN [10]. Therefore, the effects of type I IFN may vary in duration and intensity [9].
IFN induce activation of defense mechanisms and prepare cells for possible viral invasion. Induction of IFN production is closely associated with PRR activation. Generally, a cell first synthesizes IFN-β in response to signs of infection. Activation of the transcription factors NFκB and IRF-3 is required to this end. IFN-β stimulates the production of other IFN through its autocrine action associated with activation of IRF-7. IRF-7, in turn, binds to IFN-β and IFN-α gene promoters, enhancing the synthesis of those cytokines [3][11] [3,11].
Type I IFN interact with the heterodimeric IFNα receptors. Ligand binding causes dimerization of receptor subunits and activation of tyrosine kinases JAK1 and Tyk2, which phosphorylate the transcription factors STAT1 and STAT2 [3][12][3,12]. Due to interaction with IRF-9, the ISGF3 heterotrimeric complex is formed. The complex is bound by the ISRE regulatory element that is located on the promoters of most ISG. Consequently, type I IFN enhance the transcription of hundreds of genes and contribute to the cell’s antiviral response [3][13][3,13]. It is important to note that the expression of an entire ensemble of genes is necessary to limit viral replication; expression of single genes alone is not capable of providing a sufficient antiviral response [3]. The signaling pathways that are affected by the actions of type III IFN are generally similar to those of type I IFN [14].
It has been shown that other signaling pathways can affect the induction of IFN-dependent transcription. The JAK-STAT signaling pathway is known to be associated with the phosphotidylinositol-3-dependent signaling cascade [12][15][12,15]. Moreover, the effects of type I IFN are associated with increased activity of MAPK-dependent signaling cascades [3][16][3,16]. In addition to direct antiviral effects, type I IFN have immuno-modulatory properties [3][9][17][18][19][3,9,17,18,19].
Cells have mechanisms for inhibiting the effects of type I IFN. These mechanisms may be focused on the attenuation of JAK-STAT-dependent signaling cascades. For example, it has been shown in vivo that injections of IFN-α cause activation of negative regulators, such as SOCS-1 and SOCS-3 [20][21][20,21]. Moreover, a prolonged effect of the USP18/UBP43 inhibitor ISG-encoded isopeptidase has been described [20]. The USP18/UBP43 protein is an ISG15-specific protease [22]. Knockout of USP18/UBP43 in mice results in hypersensitivity to type I IFN. This protein inhibits JAK-STAT signaling pathways by binding to the IFNαR2 subunit and blocking the interaction between the JAK1 kinase and the heterodimeric receptor [23]. Pre-incubation with type I or type III IFN has been shown to cause desensitization of cells to the stimulatory effects of IFN-α. The degree of desensitization depends on the expression level of USP18/UBP43. Thus, there is a negative feedback loop attenuating both the type I and type III IFN [9].
2. IFN-λ Play a Distinct Anti-Viral Role in Collaboration with Other IFN
2.1. IFN-λ Structure
Type III IFN (IFN-λ) are a group of cytokines that is related to type I IFN and elicit similar antiviral effects [1][24][1,24]. Four IFN-λ subtypes have been found in humans: IFN-λ1 (IL-29), IFN-λ2 (IL-28A), IFN-λ3 (IL-28B), and IFN-λ4. All of these proteins are encoded on the 19th chromosome, and these genes consist of five or six exons [7]. Several of these IFN (λ1, λ2, λ3) feature a high degree of amino acid conservation [25][26][25,26], which suggests the presence of a single ancestor [7][27][7,27]. IFN-λ2 and IFN-λ3, for example, are 96% identical in amino acid sequence; they differ by just seven residues. However, IFN-λ1 is only 80% identical to them in primary structure and also differs in disulfide bond configuration; IFN-λ1 does not form a third disulfide bond [4][25][4,25]. Initially, IFN-λ4 was thought to be a pseudogene. However, it is now known that humans have the IFNL4 gene, but in some populations, there is a polymorphism (ss469415590, TT/ΔG). The TT allele causes a frameshift leading to suppression of IFNL4 production, while the ΔG allele results in the functional IFNL4 gene [4][28][4,28]. Although the product of this gene is only 40.8% identical to IFN-λ3 [29], IFN-λ4 interacts with the heterodimeric receptor common to all IFN-λ. It has been revealed that N-glycosylation of IFN-λ4 is necessary for protein secretion. IFN-λ1 is also known to have a potential N-glycosylation site at asparagine residue 65 [4]. Only IFN-λ2 and IFN-λ3 have been found in mice, and murine IFN-λ1 is a pseudogene [26][30][31][26,30,31]. Murine IFN-λ2 and IFN-λ3 are also N-glycosylated [27]. IFN-λ are conserved in tetrapod vertebrates; and tetrapod type III IFN form a monophyletic group [32]. IFN-λ are considered to be derived from “IL-10-like” cytokine family [33]. Fundamental similarities between type III IFN systems of mammals and birds indicate that type III IFN might play a significant role in defending mucosal surfaces against viral infections in birds [34]. Two groups of IFN were discovered in fishes, but fish IFN are evolutionarily closer to type I than to type III tetrapod IFN [33].
Although type III IFN (IFN-λ) should be considered most closely related to type I IFN (according to primary structure), IFN-λ are close to IL-10 (and other “IL-10-like” cytokine family members) in their spatial structure [25][26][25,26]. The spatial structure of IFN-λ includes five α-helices (A, C, D, E, F) and an element B having a less-defined structure [4]. Helix A, helix F, and the AB loop are responsible for the interaction between lambda IFN and their receptors. Certain amino acids located in the AB loop (Lys49 and Arg51 for IFN-λ3; Arg49 and His51 for IFN-λ2) have a critical effect on affinity for the IFNλR1 subunit of the receptor. Helix D is responsible for binding to the IL-10R2 subunit (Gly95 for IFN-λ3, and Val95 for IFN-λ2, are considered to be important for the interaction). Lambda IFN differ in their receptor subunit affinities; the stabilities of their final ligand-receptor complexes also differ [29]. Hepatitis C virus (HCV) persistence is strongly associated with the expression of a functional IFNL4 gene, whereas the nonfunctional IFNL4 gene is associated with more rapid viral clearance [28]. Humans have several mechanisms to limit the expression of functional IFN-λ4 through noncoding splice variants and nonfunctional protein isoforms. Moreover, protein-coding IFN-λ4 mRNA are not loaded onto polyribosomes and lack a strong polyadenylation signal, which results in poor translation efficiency [35]. Amino-acid substitution (P70S) is also strongly associated with HCV clearance. Patients harbouring the S70 variant display lower ISG expression, better treatment response rates and better spontaneous clearance rates, compared with patients coding for the fully active P70 variant [36]. Interestingly, variant E159 (E159K substitution) of IFN-λ4, that was found in some ancient African populations, exhibit more significant antiviral activity than wild-type IFN-λ4. Thus, substitution E154K also negatively affects IFNλ4 activity by reducing its secretion and potency [37].
2.2. Expression of IFN-λ
Synthesis of type III IFN is induced by viral infection and PRR activation (TLR, RLR, Ku70), and it occurs in various tissues. For example, high IFN-λ levels are observed in the lungs and liver [25][26][38][25,26,38]. Many cell types are capable of producing both IFN-α and IFN-β (IFN-α/β) with IFN-λ, but there are exceptions. For example, in response to infection with influenza virus or herpes simplex virus type-2 (HSV-2), macrophages can produce only IFN-α/β, but not IFN-λ [30][39][30,39]. Infection with swine influenza virus (H3N2) lead to IFN-β, but neither IFN-α nor IFN-λ1 expression in porcine macrophages [40]. IFN-λ expression has been discovered in respiratory epithelial cells, keratinocytes, dendritic cells, hepatocytes, and primary neuronal cells [30]. Moreover, IFN-λ are the most common IFN produced by respiratory epithelium in response to dsRNA (poly(I:C), a TLR-3 agonist); agonists of other TLR do not induce production of IFN-α/β or IFN-λ in this cell type [38]. A potent IFN-λ response is observed upon infection of human respiratory epithelial cells with respiratory viruses, such as influenza or rhinovirus [30][38][39][40][41][42][30,38,39,41,42]. Additionally, swine influenza virus (H3N2) up-regulates IFN-λ1 in porcine epithelial cells as well as in precision-cut lung slices [40]. However, myeloid dendritic cells (mDC) and plasmacytoid dendritic cells (pDC) appear to be the major producers of IFN-λ [43]. It was shown that CpG DNA (a TLR-9 agonist) induces the expression of IFN-α/β and IFN-λ in pDC, while lipopolysaccharides (LPS) and poly(I:C) (TLR-4 and TLR-3 agonists, respectively) induce expression of IFN-β and IFN-λ1-3 in mDC (but not IFN-α) [29].
2.3. Molecular Mechanism of IFN-λ Induction
IFN-λ are induced by pathways and factors similar to those involved in the induction of IFN-α/β. Moreover, IFN-λ production is mediated by activation of the same PRRs as IFN-α/β [26]. For example, expression of IFN-λ is significantly mediated by activation of the RIG-I and MDA-5-dependent signaling pathways in respiratory and dendritic cells [5][44][45][5,44,45]. However, it was found that the production of IFN-λ1, but not type I IFN, can also be induced by the activation of certain DNA sensors (Ku70). Induction of IFN-λ1 synthesis, in this case, is mainly associated with activation of the IRF-1 and IRF-7 transcription factors [46].
It was shown that the transcription factors IRF-1, IRF-3, IRF-7, and NFκB can bind to the promoters of IFNL genes. The synergistic effect of these transcription factors allows for maximum induction efficiency [45]. However, it is worth noting that expression of IFN-λ2 and IFN-λ3, like IFN-α, is still predominantly regulated by IRF-7 and NFκB; expression of IFN-λ1, like IFN-β, requires the combined action of IRF-3, IRF-7, and NFκB [47]. In mice lacking IFN-λ1, IRF-3 is not involved in the induction of IFN-λ expression in response to metapneumovirus infection [5][44][5,44]. Additionally, the Med23 subunit of the eukaryotic multiprotein mediator, which interacts with transcription factors and RNA polymerase II, binds directly to IRF-7 and induces IFN-λ synthesis. However, Med23 is not able to enhance the IRF-7-mediated induction of IFN-β transcription. These data emphasize an additional selectivity of the IFN response [48]. A detailed review of type I and type III IFN induction mechanisms, and their differences, has been published [26].
Expression kinetics for IFN-λ depend on cell type and induction conditions. In PBMC and fibroblasts, it has been shown that peak IFN-λ3 expression occurs 24 h after infection with cytomegalovirus, while the IFN-λ1 peak is 6 h after infection [49]. When primary human hepatocytes are infected with HCV, increased IFN-λ4 mRNA levels can be detected 2–4h after infection. However, the expression subsides after 8 h, which may suggest either: absence of a positive-regulation feedback loop; or (conversely) induction of specific negative-feedback mechanisms [29]. There is limited information about IFN-λ negative regulation (reviewed in [50]). Stimulation by type III IFN leads to ISG expression that includes SOCS and IL-10 expression. Excessive SOCS-1 expression is associated with reduced STAT1 phosphorylation as well as reduced ISG expression [51]. Type III IFN activity may be inhibited in the presence of IL-10 [52]. Additionally, it should be noted that the level of IL10R2 subunit is modulated by ubiquitination leading to degradation of nonspecific subunits [53].
2.4. The IFN-λ Receptor (IFNλR)
IFN-λ actions are carried out through the heterodimeric IFNλR, consisting of the IFNλR1 and IL10R2 subunits [25][54][25,54]. The IL10R2 subunit is also part of the receptor complexes for IL-10, IL-22, and IL-26; it is expressed in cells of various tissues. Interestingly, the IFN-λ1 and IL-10-like cytokines bind with low affinity to the IL10R2 subunit itself. In turn, IFN-λ1 specifically binds to the IFNλR1 subunit in a 1:1 stoichiometric ratio [25]. An IFNλR has also been found in mice; the murine IFNλR amino acid sequence is approximately 67% similar to the human. It should be noted that both murine and human IFN-λ act non-specifically in terms of host species: murine IFN-λ can bind to human IFNλR [27]. On the other hand, murine and human IFN-λ exhibit some species specificity. For instance, murine IFN-λ3 is 51 times less active in human A549 cells than in mouse LKR10 cells. However, IFN-λ4 is more active in mouse cells [55].
Expression of the IFNλR1 subunit demonstrates restricted cellular distribution. For example, IFN-λ does not act on fibroblasts, splenocytes, macrophages, or (migrated, bone marrow-originating) endothelial cells, since IFNλR1 is not expressed in these cells, while IFN-α is able to activate all of them [27][30][27,30]. High IFNλR1 expression has been found in the lungs, intestines, liver, and upper epidermis [30]. Expression of IFNλR1 is mainly restricted to: epithelial cells [11], keratinocytes [56], differentiated dendritic cells (pDC and mDC) [57][58][57,58], and hepatocytes [59]. In monocytes and B cells, low levels of IFNλR1 expression are detected. Thus, these cells respond extremely weakly to IFN-λ [60] [60]. As such, mucous membranes of the respiratory and gastrointestinal tracts are the primary tissue targets of IFN-λ [11]. This tissue specificity correlates with IFN-λ antiviral activity, which is seen mainly with viruses featuring high tropism for epithelial cells, like Orthomyxoviridae, Pneumoviridae, Coronaviridae, Picornaviridae, Herpesviridae, Flaviviridae, Reoviridae, Arenaviridae, Caliciviridae (Table 1. Viruses affected by IFN-lambda (Type III interferons)) [30]. In general, type III IFN control infection at mucosal barrier sites, while type I IFN are important for broad systemic infection control.
Table 1. Viruses affected by IFN-lambda (Type III interferons).
| Virus Family | Common Names |
Virus Genome | Infected Cells Expressing IFN-λ and IFN λR | Effects of IFN-λ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Myxoviridae | Influenza A Influenza B |
− strand ssRNA | Respiratory epithelia, keratinocytes, mDC and pDC, hepatocytes and primary neuronal cells; NOT macrophage | IFN-λ decreases influenza virus replication in a dose-dependent manner in respiratory and gastrointestinal epithelial cells by up-regulating ISG ( | MX1 | , | OAS | , | IFITM1) [61]. IFN-λ is more anti-proliferative and anti-inflammatory than IFN α/β. Anti-proliferative effects due to up-regulation of p53 can increase susceptibility to bacterial pathogens [62]. | ) [61]. IFN-λ is more anti-proliferative and anti-inflammatory than IFN α/β [62]. Anti-proliferative effects due to up-regulation of p53 can increase susceptibility to bacterial pathogens [62]. |
| Paramyxoviridae | Resp. syncitial virus (RSV); Metapneumovirus Measles virus |
− strand ssRNA | Respiratory epithelia | Mice treated with IFN-λ2 and -λ3 had decreased viral titers, less pulmonary inflammation, and higher survival rates [5]. Metapneumovirus replication is attenuated in DC through MDA-5-mediated IFN response [44]. IFN-λ restricts measles replication in lung epithelial cells [63]. |
||||||
| Arenaviridae | Lymphocytic choriomeningitis virus (LCMV) |
− strand ssRNA | Respiratory epithelia, DC | IFN-λ2 and -λ3 elicit an antiviral effect against LCMV in lung cell culture [64]. | ||||||
| Flaviviridae | Hepatitis C Dengue virus |
+ strand ssRNA | Primary hepatocytes DC and Lung epithelial cells |
Successful Heptitis C treatment is associated with human genetic SNPs in IFN-λ3 promoter [65] and in IFN-λ4 [28]. IFN-λ1 reduced DC migration by reducing CCR-7 expr [66]. IFN-λ1 and -λ2 increase antiviral ISG (OAS and Mx1) and thus decrease virus loads [67]. |
||||||
| Caliciviridae | Norovirus (NoV) |
+ strand ssRNA | Intestinal epithelia | IFN-λ clears persistent NoV, affects gut microbiota, and prevents transmission of acute NoV [68]. | ||||||
| Picornaviridae | Rhinovirus |
+ strand ssRNA | Respiratory epithelia, DC | IFN-λ decreases rhinovirus replication and the asthmatic effects of rhinovirus. In a murine model for asthma, treatment with IFN-λ2 reduces Th2, eosinophils and neutrophils in bronchial fluid [30][41][42]. | IFN-λ decreases rhinovirus replication and the asthmatic effects of rhinovirus. In a murine model for asthma, treatment with IFN-λ2 reduces Th2, eosinophils and neutrophils in bronchial fluid [30,41,42]. | |||||
| Picornaviridae | Coxsackie virus |
+ strand ssRNA | Primary human hepatocytes | Coxsackie titers were 10–100X lower in IFN-λ-treated cells [69]. | ||||||
| Coronaviridae | MERS-CoV SARS-CoV-1 and -2 |
+ strand ssRNA | Respiratory epithelia | The coronaviruses induce little type I or type III IFN, but treatment with PEGylated IFN-λ1 decreased SARS-CoV-2 titers [70]. | ||||||
| Herpesviridae | Cytomegalo- virus (CMV) Herpes (HSV-1 HSV-2) |
dsDNA | CMV infects human foreskin fibroblasts. HSV infects buccal or genital mucosa |
IFN-λ3 lowers infection with CMV [49]. IFN-λ lowers infection with HSV-1 [48]. |
||||||
| Hepadnaviridae | Hepatitis B (HBV) |
+ strand ssDNA | HBV infects primary hepatocytes. | HBV infection up-regulates expression of IFN-λ [26]. IFN-λ1 significantly reduced viral load during infection with HBV [71]. |
2.5. The Effects of IFN-λ on Cells
Lambda IFN are secreted into the extracellular space and exert autocrine or paracrine effects by binding to cell surface receptors. Although IFN-α/β and IFN-λ actions are realized through different receptors, they lead to the activation of similar signaling pathways. Upon IFN-λ binding, receptor subunits dimerize leading to activation of JAK/STAT-dependent signaling pathways: activation of JAK1 and Tyk2 tyrosine kinases; phosphorylation of receptor subunits; recruitment and subsequent phosphorylation of STAT1 and STAT2 proteins, and to a lesser extent STAT3-STAT5; and formation of the ISGF3 transcription complex. Interestingly, STAT1 can also be activated by the actions of various cytokines, while STAT2 phosphorylation is caused by the specific action of type I or type III IFN [72]. The ISGF3 complex is also formed in response to the actions of type I IFN. Therefore, IFN-λ functions significantly overlap with type I IFN functions and cause the expression of similar ISG. In addition to activating JAK/STAT signaling cascades, IFN-λ also influences MAPK signaling pathways, including the Erk, Jnk, and p38 kinases [14][26][73][74][14,26,73,74].
There are some differences in the mechanisms activated by type I and type III IFN. In intact cells, IFN-λ activate STAT-dependent signaling pathways slightly more weakly than IFN-α, which is associated with higher basal type I IFN levels. Interestingly, the JAK2 kinase (necessary for phosphorylation of STAT1) is specifically activated in response to IFN-λ; this may underlie the differences in IFN-λ and IFN-α/β effects. Moreover, gene knockout of JAK2, or its inhibition by substances (such as AG490 or 1,2,3,4,5,6-hexabromocyclohexane), can specifically block IFN-λ-dependent signaling cascades without affecting type I IFN-depended signaling pathways [75].
2.6. Immuno-Modulatory Activity of IFN-λ
IFN-λ-mediated signaling also regulates the immune response. The presence of phosphorylated STAT-3, STAT-4, and STAT-5 forms indicates the existence of an additional level of complexity. Thus, there are several associations: STAT3 phosphorylation is also a signaling mechanism used by members of IL-10-like cytokines (IL-10, IL-19, IL-20); phosphorylated forms of STAT5 are often associated with IL-2, IL-3, and GM-CSF; and STAT4 is associated with a T helper type 1 (Th1)-mediated immune response. In general, an entire body of knowledge indicates the presence of additional immuno-modulatory activities of IFN-λ [52].
In early studies, it was shown that IFN-λ1 causes the secretion of IL-6, IL-8, and IL-10 in PBMC. Selective blocking of IL-10 with specific antibodies leads to a decrease in the required dose of IFN-λ1 for the secretion of IL-6. In turn, the addition of IL-10 reduced the effects of IFN-λ1. Therefore, the existence of a feedback mechanism can be assumed by which IFN-λ1 causes the secretion of IL-10, which inhibits the effects of the former. This mechanism may be associated with competition between IFN-λ1 and IL-10 for binding to the IL10R2 subunit [76]. The IL-22 receptor also contains the IL10R2 subunit. IL-22 acts synergistically with IFN-λ and causes activation of STAT1-dependent signaling pathways in the suppression of rotavirus infections [77]. Dendritic cells express IFNλR during differentiation from monocytes. The dendritic cells, maturing upon stimulation with IFN-λ, induce IL-2-dependent proliferation of a population of CD4+/CD25+/Foxp3+ regulatory T cells [74].