The cardiac IKs (KCNQ1/KCNE1) channel is one of the main contributors to the repolarizing currents that regulate the ventricular action potential duration (APD) and thus the QT interval in the electrocardiogram. Mutations in cardiac KCNQ1/KCNE1 channels are the most common cause of congenital defects that cause long QT syndrome (LQTS).
- KCNQ1/KCNE1
- voltage clamp
- K channels
- PUFA
- IKs
- PIP2
- Ion channles
Note:All the information (including the title) in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
The KCNQ1/KCNE1 channel is a member of the super family of voltage-gated ion channels. Voltage-gated ion channels are a class of transmembrane proteins that are activated by membrane potential and play a crucial role in regulating cellular excitation in diverse cell types including cardiomyocytes and neurons. For example, in cardiomyocytes, voltage-gated Na+ (Nav), K+ (Kv) and Ca2+ (Cav) channels are necessary for the initiation, maintenance, propagation and termination of action potentials [1,3,9][1][2][3].
The KCNQ1/KCNE1 channel consists of the alpha-subunit KCNQ1 and the beta-subunit KCNE1 [10,11][4][5]. Four KCNQ1 subunits form a Kv channel that is modulated by 1–4 KCNE1 subunits [12,13][6][7]. KCNE1 modulation is crucial for the KCNQ1/KCNE1 channel function, but the mechanisms by which KCNE1 interacts with KCNQ1 and thus modulates KCNQ1 are not fully clear yet. Like other Kv channels, the KCNQ1/KCNE1 channel contains both a voltage-sensing domain (VSD) and a pore domain (PD) [14][8]. Upon activation, the positively charged voltage sensor of VSD senses the membrane potential change and moves outwards within the membrane, opening the pore through the VSD–PD coupling [15–17]9][10][11]. Several modulators can regulate the KCNQ1/KCNE1 activity. They can either activate or inhibit the KCNQ1/KCNE1 channel. Some modulators, such as PUFA analogs[12][13][14] [18–20] and chromanol 293B [21–23][15][16][17], are antiarrhythmic in that they can regulate the APD in cardiomyocytes and the QT interval in animals by modifying the KCNQ1/KCNE1 activation.
2. KCNQ1/KCNE1 Channel Role in the Cardiac Action Potential
The heart is a blood pump whose activity is controlled by cardiac electrical activity [1]. During each heartbeat, a healthy heart has an orderly progression of action potentials that start with the sinoatrial node, then spread out through the atrium, pass through the atrioventricular node down into the Purkinje fibers and finally spread out through the ventricles. The cardiac electrical activity can be detected by the electrocardiogram, also called ECG or EKG [2][18]. In a normal ECG recording, each heartbeat contains five different waves: P, Q, R, S and T waves (Figure 1A) [2][18]. The QT interval is measured from the beginning of the Q wave to the end of the T wave. Patients with a prolonged QT interval are likely to be clinically diagnosed as having long QT syndrome, which can lead to Torsades de pointes, ventricular fibrillation and sudden cardiac death [3,4,24][2][19][20]. Conversely, patients with a shortened QT interval might be diagnosed as having short QT syndrome (SQTS), which can lead to atrial fibrillation (AF) [3][2].
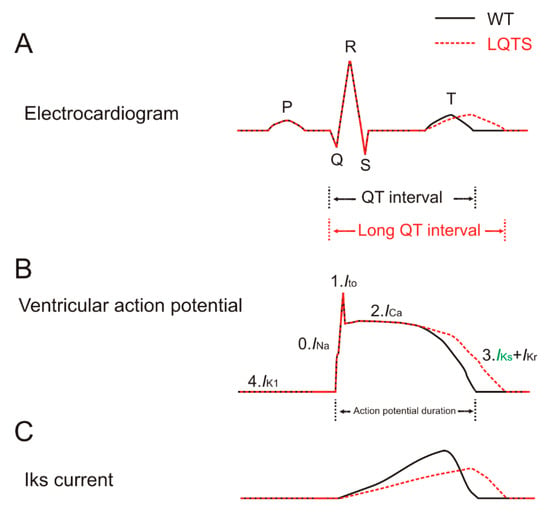
Figure 1. Long QT syndrome (LQTS) caused by a loss of the IKs current. Normal electrocardiogram, ventricular action potential and IKs current are depicted by black solid lines. The electrocardiogram, ventricular action potential and IKs current associated with LQTS are depicted by red dashed lines. Horizontal and vertical axes are not shown for clarity. (A) The electrocardiogram consists of five waves: P, Q, R, S and T waves. The P wave represents the depolarization of the atria. The QRS complex represents the rapid ventricular depolarization. The T wave represents the repolarization of the ventricles. The QT interval is the interval from the beginning of the Q wave to the end of the T wave. Patients with a prolonged QT interval can cause LQTS. Action potential duration is indicated. (B) The ventricular action potential consists of five phases, phase 0 to phase 4. Phase 0 is the depolarization phase that is mediated by an inward Na+ current. Phase 1 is mainly mediated by a transient outward K+ current. Phase 2 is the plateau phase that is mainly mediated by an inward Ca2+ current and outward K+ current. Phase 3 is the repolarization phase that is mainly mediated by outward K+ (IKs in green and IKr) currents. Phase 4 is mainly mediated by an outward K+ current (IK1). (C) The IKs current underlies the repolarization of the ventricular action potential, shown as phase 3 in (B).
The ventricular cardiac action potential is mainly mediated by voltage-gated Na+, Ca2+ and K+ channels (Figure 1B) [3][2]. These channels are closed at the negative diastolic membrane potential but open upon membrane depolarization during systole. There are five phases of the ventricular action potential, phase 0 to phase 4 (Figure 1B) [1,3,25][1][2][21]. Phase 0 starts when the Nav channel activates, leading to the influx of Na+ ions from the outside of the membrane, which causes a strong depolarization. During phase 1, the Nav channel rapidly inactivates while a specific Kv channel activates, leading to a transient outward K+ current (Ito). The inactivation of Nav and the activation of Ito lead to a small hyperpolarizing notch in the membrane potential. Phase 1 is followed by phase 2 when the Cav channel (L-type Ca2+ channel) activates. The current is now mainly mediated by the influx of Ca2+ ions, which contributes to the sustained depolarization or plateau phase of the ventricular action potential. Phase 3 begins when enough of the delayed rectifier Kv channels (IKs and IKr) are activated, causing a more outward K+ current than inward Ca2+ and Na+ currents. This process leads to the repolarization of the membrane potential and hence the termination of the action potential. The IKr and IKs contribute to the fast and slow components of the delayed rectifier K+ currents, respectively. A prolonged repolarization of the action potential caused by a loss of the IKs or IKr current is the most common cause for congenital LQTS-caused Torsades de pointes, ventricular fibrillation and sudden cardiac death (Figure 1C) [3,4][2][19]. For instance, loss-of-function mutations in IKs channels cause LQT1 (mutations in the KCNQ1 subunit) and LQT5 (mutations in the KCNE1 subunit) by reducing the amplitude of the repolarizing outward IKs current and thus increasing the APD [5,6][22][23]. Curiously, one KCNQ1 mutation was recently found to cause sever LQTS by reducing trafficking of IKr channels [26][24]. Conversely, rare severe gain-of-function mutations in IKs channels cause SQTS and AF by increasing IKs channel activity and thereby shortening the APD [3,27][2][25]. IKs channels have been shown as important for the repolarization process of both atrial and ventricular action potentials, but more important in the ventricular action potentials. Instead, the ultrarapid delayed rectifier IKur channel, only expressed in the human atria, is the predominant delayed rectifier current responsible for the atrial repolarization and is related to the most leading cause of AF [28][26]. In phase 4, the inwardly rectifying K+ (IK1) channel is open to set the diastolic membrane potential around −90 mV.
3. Architecture of KCNQ1/KCNE1 Channels
The KCNQ1/KCNE1 channel consists of two subunits: the pore-forming subunit KCNQ1, also known as Kv7.1 or KvLQT1, and the auxiliary subunit KCNE1, also known as MinK [10,11] [4][5](Figure 2).
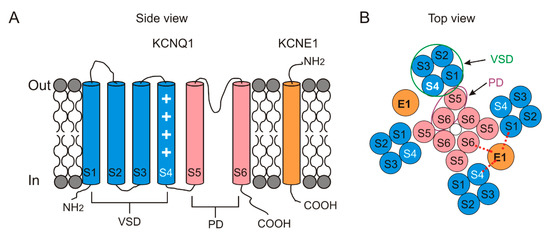
Figure 2. Topology of KCNQ1 and KCNE1. In the KCNQ1 subunit, S1–S4 transmembrane segments form the peripheral voltage-sensor domain (VSD) in blue and S5–S6 transmembrane segments form the central pore domain (PD) in pink. The KCNE1 subunit is one single transmembrane segment in orange. (A) Schematic side view of one KCNQ1 subunit and one KCNE1 subunit. The white plus symbol in S4 represents the positive gating charges. (B) Schematic top view of a tetrameric KCNQ1 channel with only two KCNE1 subunits. The number of the KCNE1 subunit varies from 1 to 4. KCNE1 is located in between S1, S4 and S6 from KCNQ1. Red dashed line indicates putative interactions between KCNE1 and S1, S4 and S6 from KCNQ1. A VSD (green circle) from one subunit is adjacent to a PD (purple oval) from its neighboring subunit.
3.1. KCNQ1
The KCNQ1 subunit itself expressed alone can form the tetrameric KCNQ1 channel (Figure 2B). KCNQ1 belongs to the family of KCNQ potassium channels, consisting of five members: KCNQ1–KCNQ5 [29][27]. The KCNQ1 channel is widely expressed in various tissues including the heart, inner ear, pancreas, kidney and brain [14,29–32][8][27][28][29][30]. KCNQ1 expressed alone elicits a fast activating current (relative to KCNQ1/KCNE1 currents, Figure 3A), undergoing a rapid inactivation, which can be seen in the hooked tail currents [33,34][31][32].
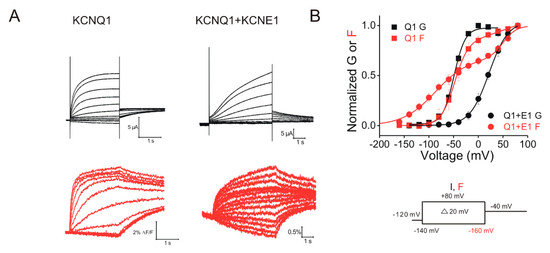
Figure 3. Voltage-clamp fluorometry recordings of KCNQ1 and KCNQ1/KCNE1 channels. (A) Representative current (black) and fluorescence (red) traces from KCNQ1 and KCNQ1/KCNE1 channels in response to the indicated voltage protocol (right). Cells are held at −120 mV and stepped to voltages between −140 (−160 mV for fluorescence) and +80 mV in +20 mV followed by a step to −40 mV. (B) Voltage dependence of currents (black) and fluorescence (red) from KCNQ1 (squares) and KCNQ1/KCNE1 (circles) channels. KCNE1 shifts the voltage dependence of the current activation of KCNQ1 to more positive voltages and separates the voltage sensors movement of KCNQ1. This suggests that S4 in KCNQ1/KCNE1 moves in two steps.
The KCNQ1 channel, like most other Kv channels, is composed of four subunits [14][8]. Each subunit contains six transmembrane segments, S1–S6 (Figure 2). The S1–S4 segments in each subunit form a peripheral voltage-sensing domain (VSD), while the S5 and S6 segments form a pore domain (PD). Four PDs together form the centrally located K+-conducting pore. The S4 segment harbors several positively charged residues and therefore senses the voltage changes across the membrane (Figure 2A). Like many Kv channels (but in contrast to IKr channels), KCNQ1 channels have a domain-swapped structure in which the VSD from one subunit is adjacent to the PD from the neighboring KCNQ1 subunit.
Ground-breaking high-resolution structures of many voltage-gated ion channels have been recently reported as advances have occurred in cryo-electron microscopy (cryo-EM) techniques. These structural studies have shed light on the structure–function relation of many ion channels. The Xenopus and human KCNQ1 structures in complex with Calmodulin (CaM) were revealed by MacKinnon’s lab using cryo-EM [35,36][33][34]. Generally, voltage sensors of most Kv channels are in the resting conformation at negative voltages while in the activated conformation at positive voltages [37][35]. However, some Kv channels, such as KCNQ1 [34[32][36],38], Shaker[37] [39] and Kv1.2 [40][38], have been shown to also exhibit an intermediate conformation of voltage sensors [39–41][37][38][39]. Since structures of KCNQ1-CaM mentioned above were resolved at 0 mV, the voltage sensor of those channels was proposed to be in the activated conformation. To elucidate the voltage-sensing mechanism of KCNQ1 and likely other Kv channels, structures of voltage sensors at the intermediate conformation and resting conformation are needed. Recently, a nuclear magnetic resonance (NMR) structure of a human KCNQ1 VSD in an intermediate state[39] [41] has been reported, although the PD is missing in this structure. This intermediate VSD conformation was suggested to be different from the cryo-EM activated VSD conformation using site-mutated mutagenesis and voltage-clamp fluorometry (VCF) [35][33]. Over the years, resolving the three-dimensional resting-state structure of VSD has been challenging in voltage-gated ion channels because the resting state predominates only at very negative voltages. Recently, Catterall and his colleagues[40] [42] presented a cryo-EM structure of a bacterial Nav channel in the resting state. The VSD was captured in the resting state by introducing some voltage-shifting mutations and a disulfide crosslink (between G94C in S4 and Q150C in S5) to stabilize the resting state of S4. S4 was shown to move vertically ~11.5 Å with a significant rotation from the resting state to the activated state. Although this is the resting-state structure of a Nav channel in prokaryotes, it helps to understand how the voltage sensor moves and how it couples to the channel opening in voltage-gated ion channels. Maybe it will also be possible to reveal the resting VSD structure of KCNQ1 channels by using voltage-shifting mutations and disulfide crosslinks to trap the VSD in the resting state.
3.2. KCNE1
KCNE1 belongs to the family of KCNE auxiliary subunits. In total, there are five members of the KCNE family, labeled KCNE1–KCNE5. All of the members of the KCNE family are single-transmembrane segment proteins (Figure 2A) that differentially modify the properties of KCNQ channels in diverse tissues [14][8]. We here only focus on the KCNE1 modulation of KCNQ1 in the heart.
KCNE1 modifies the KCNQ1 channel function in several ways. For example, co-expression of KCNE1 slows the activation kinetics, increases the voltage-dependent current amplitude and shifts the voltage dependence of activation to more positive voltages of KCNQ1 channels in heterologous expression systems (Figure 3) [10,11][4][5]. This slowing of the activation kinetics is very important for generating the slowly activating IKs currents that regulate the APD and QT interval. Furthermore, some studies have shown that KCNE1 induces a larger single-channel conductance in KCNQ1 channels [43–45][41][42][43]. These studies suggested that the increase in the macroscopic current in the presence of KCNE1 is, at least partly, due to the increased apparent single-channel conductance of the KCNQ1 channel. KCNE1 has also been suggested to alter the ionic selectivity and to eliminate the inactivation of the KCNQ1 channel [33,46][31][44]. Conti et al.[44] [46] found that, compared to KCNQ1 channels, KCNQ1/KCNE1 channels display a significantly lower Rb+/K+ permeability ratio. They further proposed that the Rb+/K+ permeability ratio is associated with the inactivation in KCNQ1 channels [47][45], yet the molecular mechanism underlying the relation between the Rb+/K+ ratio and the inactivation is not fully understood.
The structure of KCNQ1/KCNE1 has not been obtained yet. However, a recent cryo-EM structure of the KCNQ1/KCNE3-CaM complex gives us a hint about how the KCNE1 subunit interacts with KCNQ1 and modulates the KCNQ1 activity [35][33]. Unlike KCNE1, KCNE3 dramatically shifts the voltage dependence of KCNQ1 channels to more negative voltages and thus makes KCNQ1/KCNE3 channels constitutively open in the physiological voltage range (from −90 to +50 mV) [48,49][46][47]. Comparing the structures between KCNQ1-CaM and KCNQ1/KCNE3-CaM, Mackinnon et al.[33] [35] found that KCNE3 lies in between S1, S4, S5 and S6 from three different subunits, suggesting KCNE1 and KCNE3 might share a similar location when associated with KCNQ1.
3.3. Interaction between KCNQ1 and KCNE1
The stoichiometry of KCNQ1 and KCNE1 has been a long-lasting debate. Some studies have shown that KCNQ1 subunits and KCNE1 subunits together form the KCNQ1/KCNE1 channel with a flexible stoichiometry from 4:1 to 4:4 in heterologous expression systems [12,13][6][7]. Other evidence from experiments using single-molecule bleaching approaches indicates that the human surface KCNQ1/KCNE1 channel contains four KCNQ1 subunits and only two KCNE1 subunits (Figure 2B) [50][48]. The cryo-EM structure of KCNQ1/KCNE3 (a paralog of KCNE1) mentioned above supports the idea that a 4:4 stoichiometry is possible [35][33]. Therefore, the number of KCNE1 for the tetrameric KCNQ1 seems to be possible from one to four. Noteworthy, the KCNQ1/KCNE1 stoichiometry in cardiac cells has not been determined yet. In addition, other KCNE subunits are also expressed in cardiac tissues[49] [51] and could compete with KCNE1 for association with KCNQ1, potentially making KCNQ1/KCNE1 stoichiometry more complex.
How the KCNE1 subunit associates and functionally interacts with the KCNQ1 subunit remains unclear. The KCNE1 subunit has been suggested to be in direct physical contact with different sites of the KCNQ1 subunit (Figure 2B). Several studies[50][51][52] [52–54] have demonstrated that KCNE1 directly binds to the pore, particularly the S6 segment, of the KCNQ1 channel to control the KCNQ1 gating. For example, FTL residues (F57, T58 and L59) in KCNE1 have been suggested to interact with S338, F339 and F340 in S6. Our recent study[47] [49] about how KCNE1 acts on the gate of KCNQ1 supports the interaction between FTL residues and F339. In addition, the direct interaction between KCNE1 and S6 is supported by the evidence that a couple of residues (K41 and L42) from KCNE1 can form disulfide bonds with K324 in S6 when mutated to cysteines [55][53]. KCNE1 is also suggested to have contact with the S4 segment and shift the voltage dependence of the S4 movement [56][54]. Comparison of the S4 movement between KCNQ1 and KCNQ1/KCNE1 channels suggests that KCNE1 acts on S4 and separates the two components of S4 movements further in voltage dependence [49][47].
The extracellular end of the S1 segment has drawn some attention as an allosteric region of KCNQ1 gating. In this region, there is a short stretch of residues (positions 140–147) that when mutated are associated with cardiac arrhythmia [57][55]. Evidence from immunoblot and cysteine crosslinking experiments indicates that the extracellular end of KCNE1 makes state-dependent contact with the extracellular end of S1 in the KCNQ1 channel [55,57][53][55]. Tseng et al.[55] [57] found that I145C could form disulfide bonds with KCNE1 G40C and K41C in a state-dependent way. After mutating, one at a time, the first four residues flanking the extracellular ends of S1 and KCNE1 to cysteines, Kass et al.[53] [55] found that the disulfide bond can be formed between I145C and residue K41C and L42C. The direct contact between the extracellular end of S1 and KCNE1 is also supported by studies on two gain-of-function mutations (S140G and V141M) that cause short QT syndrome and atrial fibrillation. S140G and V141M greatly slow the KCNQ1/KCNE1 channel deactivation and hence increase the repolarizing K+ current in the action potential [58,59][56][57]. However, a big difference between these two adjacent mutations is that S140G slows the deactivation kinetics in the presence or absence of KCNE1, whereas V141M acts only in the presence of KCNE1. This suggests a direct interaction between V141 and KCNE1[25] [27] and also an indirect interaction between S140 and KCNE1 through the neighboring residue V141.
Taken together, several different sites, including S1, S4 and S6, of KCNQ1 could form disulfide bonds with residues 40–43 in KCNE1, suggesting that the extracellular end of KCNE1 is very flexible and engages in conformational changes during KCNQ1/KCNE1 association [27,52,55–57][25][50][53][54][55]. These electrophysiological studies are consistent with the idea that KCNE1 lies in between S1, S4 and S6 from different subunits of KCNQ1 (Figure 2B).
4. Physiological Modulators of KCNQ1/KCNE1 Channels
4.1. Protein Kinase A (PKA)
Cardiac KCNQ1/KCNE1 channels are regulated by sympathetic nervous stimulation via the activation of beta-adrenergic receptor-mediated PKA. During exercise or stress, stimulation of the sympathetic nervous system leads to a dramatically rapid heart rate. To allow the heart to have enough diastolic filling time between each heartbeat, a shortened ventricular action potential duration and a corresponding reduced QT interval in ECG recordings are necessary [72][58]. In patients with congenital LQTS, stimulation of sympathetic discharge during exercise increases the risk of tachyarrhythmias and sudden cardiac death [73,74][59][60]. The upregulated KCNQ1/KCNE1 channel activity via PKA activation was found to be important for regulating the cardiac action potential upon beta-adrenergic stimulation [72][58]. The activation of beta-adrenergic receptors increases the intracellular levels of cyclic adenosine monophosphate (cAMP) which in turn activates PKA. PKA activation then phosphorylates the KCNQ1/KCNE1 channel and enhances KCNQ1/KCNE1 function, therefore shortening the APD [72,75,76][58][61][62]. For example, Terrenoire et al. found [72][58] that PKA simulation speeds up the activation kinetics while slowing the deactivation kinetics of KCNQ1/KCNE1 channels in CHO cells. Yazawa and Kameyama[61] [75] found that both isoprenaline (a beta-adrenergic receptor agonist) and PKA increase the amplitude of KCNQ1/KCNE1 currents in guinea pig cardiomyocytes. The regulation of KCNQ1/KCNE1 by phosphorylation at S27 in the KCNQ1 N-terminus requires protein phosphatase 1 (PP1) and the A-kinase anchoring protein Yotiao [77][63]. Yotiao was suggested to bind to the C-terminus in KCNQ1 via a leucine zipper (LZ) motif [77][63]. Mutation in the LZ motif disrupts the interaction between Yotiao and the LZ motif and thus leads to LQTS. Although most studies[58][61][63] [72,75,77] have shown that upon beta-adrenergic simulation, PKA activation upregulates the activity of KCNQ1/KCNE1 channels, some groups[64][65] [78,79] showed that sustained beta-adrenergic simulation downregulates the KCNQ1/KCNE1 activity in guinea pig cardiomyocytes. The downregulation of KCNQ1/KCNQ1 might be due to the reduced KCNE1 expression mediated by exchange protein directly activated by cAMP (Epac) but not PKA. Therefore, the effect of PKA activation following acute and chronic beta-adrenergic simulation on KCNQ1/KCNE1 channels might be different, and the molecular mechanism of the difference needs to be elucidated. Furthermore, whether PKA activation alters the voltage sensor movement and/or the VSD–PD coupling has not been tested yet, which may help to understand how PKA activation modulates the KCNQ1/KCNE1 function by beta-adrenergic simulation.
4.2. Phosphatidylinositol 4,5-Bisphosphate (PIP2)
PIP2 is a phospholipid of the plasma membranes [80][66]. PIP2 was shown to activate different cardiac ion channels and transporters, while a depletion of PIP2 keeps channels and transporters inactive [80,81][66][67]. PIP2 regulation of ion channels has been suggested to keep these channels inactive during trafficking and processing of channels in intracellular membranes, which have low PIP2 levels [82][68]. In the heart, downregulation of PIP2 was suggested to prolong the ventricular action potential because cardiac KCNQ1/KCNE1 and hERG channels are sensitive to a lack of PIP2 [81][67]. According to previous structural and functional studies, PIP2 is a necessary cofactor for the KCNQ1/KCNE1 channel function and other KCNQ channels [35,83–85][33][69][70][71]. Loussouarn et al. [83][69] found that the intracellular application of PIP2 could significantly slow down the rundown of the KCNQ1/KCNE1 currents that spontaneously occurred in the excised patch-clamp recordings. The authors proposed a model in which PIP2 stabilizes the open state of KCNQ1/KCNE1 channels. Some LQTS-associated mutations (R539W and R555C) might weaken the interaction between KCNQ1/KCNE1 and PIP2 and therefore destabilize the PIP2-mediated open state of KCNQ1/KCNE1 channels [86][72]. Li et al. [84][70] found that KCNE1 increases the PIP2 sensitivity of KCNQ1 expressed alone about 100-fold, suggesting that KCNE1 is crucial for modulating the P1P2 sensitivity in KCNQ1 channels. Zaydman et al.[73] [87] showed that PIP2 is required for the coupling between the voltage-sensing domain and the pore domain, such that without PIP2 the activation of the voltage sensor is not able to induce gate opening. This PIP2-dependent VSD–PD coupling was also seen in other KCNQ channels [88][74]. Using a mutation that abolishes the potentiation effect of PIP2 on the KCNQ channels, some studies[73][74][75] [87–89] have identified a putative PIP2-binding pocket site that contains the C-terminus, A–B helix linker, S2-S3 linker and S4-S5 linker. The recent cryo-EM structure of the KCNQ1-CaM complex[33] [35] supported the idea that in the absence of PIP2, the voltage sensor still moves but the pore remains closed. However, in the presence of PIP2, PIP2 binds to the loop connecting the S4-S5 linker and the C-terminus of KCNQ1 channels, which is consistent with the mutational studies above. Upon binding, PIP2 was seen to induce a large conformational change through a 180-degree rotation of CaM and thus to open the pore. The authors proposed that other members of the KCNQ family may share a similar PIP2-mediated gating mechanism, since the binding site harbors several conserved residues [35][33]. Recently, CP1, a molecule with some resemblance to PIP2, was shown to be able to substitute for PIP2 in the VSD–PD coupling of KCNQ channels [90][76]. CP1 was able to restore the prolonged APD induced by an IKr blocker back to normal in cardiomyocytes, which indicates CP1 could be a potential therapeutic for cardiac arrhythmias.
4.3. Adenosine Triphosphate (ATP)
ATP is a major energy source in cardiomyocytes. The ATP level dramatically decreases in cardiac cells during heart failure and acute ischemia [91,92][77][78]. ATP can directly modulate the cardiac action potential and cause arrhythmias [93][79]. For example, elevated extracellular ATP was reported to trigger cardiac arrhythmias by prolonging the PR interval and partially blocking sinoatrial node activity and atrioventricular conduction in an isolated perfused rat heart [94][80]. In addition, in the electrically stimulated rat cardiomyocytes, increased extracellular ATP was shown to induce arrhythmias [95][81].
Intracellular ATP has been shown to regulate cardiac KCNQ1/KCNE1 activity. Loussouarn et al.[69] [83] showed that the spontaneous rundown of KCNQ1/KCNE1 currents in the excised patch-clamp recordings could be slowed down by the addition of PIP2 and MgATP, which underscores the importance of ATP on channel opening. Li et al. [92][78] found that elevated intracellular ATP enhances the KCNQ1/KCNE1 activation in Xenopus oocytes and shortens the APD in cardiomyocytes. On the other hand, lowered intracellular ATP reduces the KCNQ1/KCNE1 activity and prolongs the APD. Using mutagenesis and VCF, ATP was shown to bind to the C terminus of the KCNQ1 channel and is required for the pore opening but not the voltage sensor activation or the VSD–PD coupling [92][78]. Some LQTS-associated mutations were shown to reduce the KCNQ1/KCNE1 activity by affecting the ATP sensitivity of KCNQ1/KCNE1 channels. Using simultaneous patch-clamp and FRET measurement, Kienitz and Vladimirova[82] [96] found that a loss of ATP slowed the activation of KCNQ1/KCNE1 in Chinese hamster ovary (CHO) cells. In addition, since ATP depletion caused a more pronounced inhibition of KCNQ1/KCNE1 currents compared to PIP2 depletion, they proposed that intracellular ATP is a more potent modulator of KCNQ1/KCNE1 in comparison to PIP2. In KCNQ1/KCNE1 channels, the effects of PIP2 and ATP are independent of each other, although both of them are required to activate KCNQ1/KCNE1 channels. A previous study[78] [92] suggested that PIP2 and ATP have different putative binding sites and activation mechanisms in KCNQ1 channels, as well as different KCNE1 dependence.
ATP also regulates other ion channels and therefore the action potential [97][83]. For example, activation of cardiac ATP-sensitive K (KATP) channels shortens the action potential and causes arrhythmias, while suppression of KATP could prevent arrhythmias [98][84]. KATP channels activators were shown to protect the heart against ischemia and reperfusion arrhythmias [99,100][85][86]. Therefore, the KATP channel has been proposed as a target for anti-arrhythmic treatment [101][87]. Using patch-clamp, extracellular ATP inhibits the whole-cell current of ATP-sensitive K (KATP) channels [97][83]. PIP2 was shown to prevent the current inhibition of KATP channels, suggesting the important role of PIP2 in the modulation of KATP channels by extracellular ATP. Since both cardiac KCNQ1/KCNE1 and KATP channels can be regulated by PIP2 and ATP and abnormal activity of these two channels can cause arrhythmias, maybe there is a connection, such as crosstalk, between them in the progression of cardiac diseases. Indeed, a decrease in the intracellular ATP reduces the APD by activating KATP channels while prolonging the APD by inhibiting KCNQ1/KCNE1 channels, suggesting that the heart has the ability to respond in different ways, maybe in different physiological and pathological conditions, to changes in PIP2 and ATP by various ion channel regulations.
5. Pharmacology of KCNQ1/KCNE1 Channels
5.1. Agonists
To date, there have been a few known agonists for KCNQ1 and/or KCNQ1/KCNE1 channels, such as stilbenes [102[88]89],103], mefenamic acid [103[89][90],104], ML277 [105–108][91][92][93][94], phenylboronic acid (PBA) [109][95], zinc pyrithione [110][96], CP1[76] [90] and mallotoxin (MTX) [111][97].
Stilbene was one of the first activators of KCNQ1/KCNE1 to be studied years ago. Stilbenes have been shown to increase the current amplitude [102[88]89],103], slow the deactivation kinetics[88]89] [102,103] and shift the voltage dependence of current activation to more negative voltages89] [103] of KCNQ1/KCNE1 channels expressed in Xenopus oocytes. The effects of stilbene on KCNQ1 expressed alone were also tested. Stilbene showed a significantly bigger activating effect in terms of amplitude, deactivation and conductance–voltage relation in the KCNQ1/KCNE1 channel compared to KCNQ1 alone. These differences suggest that KCNE1 is involved in the activation of KCNQ1/KCNE1 channels by stilbenes. Using site-directed and deletion mutants, stilbenes were suggested to bind to the extracellular end of KCNE1 and rescue the channel gating defect by mutations in this area, such as an LQT5-associated mutant D75N [103]89]. Mefenamic acid, a fenamate compound, has been found to shift the voltage dependence to a more negative voltage and slow down the deactivation kinetics [103]89]. Similar to stilbenes, mefenamic acid might bind to the extracellular residues flanking the transmembrane segment of KCNE1. In recent work, Wang et al.[90] [104] found that mefenamic acid increases the open probability of KCNQ1/KCNE1 channels and that K41 in KCNE1 is required for mefenamic acid’s effect on KCNQ1/KCNE1 channels. The extracellular end of KCNE1 has been shown to be important for KCNQ1 and KCNE1 associations.
In a high-throughput screen, Mattmann et al.[92] [106] identified ML277 as a potent activator for the KCNQ1 channel and showed that ML277 is highly selective against other Kv channels including KCNQ2, KCNQ4 and hERG channels. This group [108][94] later found that ML277 potentiates heteromultimeric KCNQ1/KCNE1 channels but the increasing KCNE1 expression level reduced and eventually abolished ML277’s effect on KCNQ1/KCNE1 channels, indicating a competition between KCNE1 and ML277 when interacting with KCNQ1. In addition, ML277 was shown to shorten the APD in cultured human cardiomyocytes[94] [108] and guinea pig ventricular myocytes [107][93], suggesting ML277 as a promising anti-arrhythmic drug. As previously reported [109][95], PBA activates the KCNQ1/KCNE1 channel by shifting the voltage dependence of current activation to more negative voltages. Although PBA was found to inhibit other Kv channels (Shaker and hERG channels), it activates other members of the KCNQ family (KCNQ1, KCNQ2/3 and KCNQ4). Consequently, PBA derivatives more selective for cardiac KCNQ1/KCNE1 channels can be potent activators for treatment of cardiac arrhythmias.
Interestingly, some common KCNQ2-5 activators have little or no effects on KCNQ1 or KCNQ1/KCNE1 channel activation. For example, retigabine, known as the first approved anti-epileptic drug, activates the KCNQ2-5 channels that are important for neuronal excitability [88,112,113][74][98][99]. Retigabine was shown to stabilize the open state of KCNQ2-5 channels by markedly shifting the voltage dependence of current activation to more negative voltages. However, KCNQ1 and KCNQ1/KCNE1 channels are retigabine-resistant[99] [113] and the molecular mechanism is not fully clear. One possible explanation could be that KCNQ1 lacks the conserved Trp residue in other KCNQ channels that has been shown to be essential for the putative binding site for retigabine [88,114,115][74][100][101].
Taken together, although several KCNQ/KCNE1 activators have been reported, there are some limitations, including the low efficacy and the lack of specificity, that have to be overcome when thinking about the clinical use of these activators for the treatment of LQTS and cardiac arrhythmias.
5.2. Polyunsaturated Fatty Acid (PUFA)
Recently, PUFAs have drawn more and more attention as they have been demonstrated to activate KCNQ1/KCNE1 channels efficiently, making PUFAs a promising approach for treating LQTS and cardiac arrhythmias. We have shown that PUFAs and their derivatives can enhance the activation of KCNQ1 and KCNQ1/KCNE1 channels by shifting the voltage dependence of current activation to more negative voltages and increasing the maximum conductance (Figure 4C) [18,116][12][102]. Furthermore, PUFA analogs were shown to have an antiarrhythmic effect on KCNQ1/KCNE1 currents by several pieces of evidence. One piece of evidence is that PUFA analogs are able to shorten the action potential duration and stabilize rhythmic action potential firing in isolated embryonic rat cardiomyocytes treated with Chromanol 293B, which prolongs action potentials and induces arrhythmic firing [18,117][12][103]. Another piece of evidence is that PUFA analogs can restore the QT interval and APD in isolated guinea pig heart perfused with the IKr blocker E4031 to induce a prolonged QT interval [18][12]. More recently, modified PUFAs were shown to shorten the QT interval in ex vivo and in vivo guinea pig hearts [118][104]. Finally, our group studied the effects of PUFA analogs on mutants that are associated with LQTS in KCNQ1/KCNE1 channels [19][13]. These LQTS-causing mutants are located in different sites of KCNQ1/KCNE1 channels and cause LQTS by distinctive mechanisms [119][105]. We demonstrated that N-arachidonoyl taurine (N-AT), a PUFA analog, restores gating, at least partly, in all these tested LQTS mutants, suggesting N-AT could be a novel KNCQ1/KCNE1 activator for LQTS treatment.
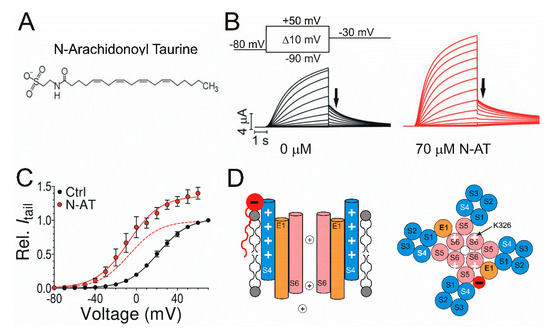
Figure 4. Effects of N-arachidonoyl taurine (N-AT) on cardiac KCNQ1/KCNE1 channels. (A) Structure of the polyunsaturated fatty acid (PUFA) analog N-AT with a negatively charged head group and a hydrophobic tail group. (B) Representative current traces from KCNQ1/KCNE1 channels in 0 μM N-AT (black) and 70 μM N-AT (red) in response to the indicated voltage protocol. Cells are held at −80 mV and stepped to voltages between −90 and +50 mV in +10 mV followed by a step to −30 mV. Arrows indicate the tail currents. (C) Voltage dependence of currents from KCNQ1/KCNE1 channels in 0 μM N-AT (black) and 70 μM N-AT (red) indicating an increased maximum conductance and negative shift of voltage dependence. Normalized voltage dependence of KCNQ1/KCNE1 channels in 70 μM N-AT is indicated as a red dashed line. (D) Illustration of the lipoelectric mechanism. Schematic side view (left) of KCNQ1/KCNE1 channels with S4 (blue), S6 (pink) and KCNE1 (orange). Electrostatic interaction between the negatively charged PUFA head group (red) and positive charges (white plus symbol) in S4. Schematic top view (right) of a tetrameric KCNQ1 channel with only two KCNE1 subunits. Electrostatic interaction between the negatively charged PUFA head group (red) and positively charged K326 (white plus symbol).
The molecular mechanism underlying the modulation of PUFAs and their analogs on the KCNQ1/KCNE1 function is fairly well understood. Structurally, PUFAs are amphipathic molecules that have both a charged hydrophilic head group and a hydrophobic tail group (Figure 4A) [120–123][106][107][108][109]. The negatively charged head group has been shown to be required for the Kv channel activation [18,123][12][109]. The negatively charged head group would electrostatically attract the positively charged S4 and enhance the S4 movement and the ensuing current activation (Figure 4D). Recently, we identified that a positively charged R231 in S4 is responsible for the electrostatic interaction between the head group and KCNQ1/KCNE1 channels [122][108]. The head group of PUFA was also suggested to electrostatically interact with the positively charged K326 in S6 to increase the maximum conductance of KCNQ1/KCNE1 channels by inducing a conformational change of the selectivity filter (Figure 4D) [122][108]. By testing PUFAs with different head groups, we found that PUFA analogs with taurine and cysteic head groups show the most pronounced activation of the KCNQ1/KCNE1 channel, suggesting that PUFAs may be developed for patients with different LQTS types [116][102].
Similar to the head group, the tail group of PUFA is necessary for the Kv channel activation [122,123][108][109]. PUFA would integrate into the plasma membrane by its hydrophobic tail group. By testing PUFAs with a carboxyl head group and different tail properties, Bohannon et al.[110] [124] found that the position of the first double bond in the tail determines the PUFAs’ effect and binding affinity to the KCNQ1/KCNE1 channel.
PUFAs have been shown to modify not only KCNQ1/KCNE1 currents but also the Nav and Cav currents underlying the cardiac action potentials. Previous studies have found that PUFAs, such as 5,8,11,14,17-eicosapentaenoic acid (EPA), inhibit the Nav currents in cultured neonatal rat ventricular myocytes[111] [125] and shorten the APD in isolated rat ventricular myocytes [126][112]. EPA was also found to suppress the L-type Cav currents in rat ventricular myocytes [127][113]. Therefore, PUFAs and PUFAs analogs are suggested to be antiarrhythmic in that they can activate KCNQ1/KCNE1 currents while inhibiting the Nav currents and Cav currents. We[14] [20] recently found that PUFAs analogs influence the activity of cardiac KCNQ1/KCNE1, Nav and Cav channels via different mechanisms. In addition, by testing PUFA analogs with different head group and with tail group properties, different PUFAs analogs display different selectivities for KCNQ1/KCNE1, Nav and Cav channels. PUFA analogs that are more selective for the KCNQ1/KCNE1 channel compared to Nav and Cav channels are able to shorten a prolonged action potential in simulated cardiomyocytes without altering other properties of the action potential. Collectively, PUFA, present in fish oil, and its analog are antiarrhythmic and potential candidates for the treatment of LQTS and cardiac arrhythmias.
5.3. Antagonist
The development of a selective KCNQ1/KCNE1 blocker is of great importance for the design of potential antiarrhythmic strategies. Several antagonists of KCNQ1/KCNE1 channels have been reported including Tetraethylammonium (TEA) ions [128–130][114][115][116], Chromanol 293B [21,22[15][16][117],131], benzodiazepine L7 [132][118], HMR 1556 [133[119][120],134], JNJ-303 [135[121][122],136], UCL2077 [137][123], XE991 [38[36][39][124],41,138], amitriptyline [139][125], Tricyclodecan-9-yl-xanthogenate (D609)[126] [140] and insulin [141][127]. Here, we only focus on a general Kv channel blocker, TEA, and a selective KCNQ1/KCNE1 channel blocker, Chromanol 293B.
The organic ion TEA has long been known to block Kv channels [142–144][128][129][130] including KCNQ1/KCNE1 channels that can be inhibited by internal TEA and inhibited weakly by external TEA [129,130][115][116]. Kurokawa et al.[116] [130] found that external TEA rapidly and reversibly blocks both the KCNQ1 and KCNQ1/KCNE1 channels expressed in CHO cells. As an open channel blocker, TEA was suggested to bind to the extracellular loop of the outer pore of the KCNQ1/KCNE1 channel, which is consistent with the common TEA-binding site in other Kv channels [143,144][129][130]. By testing the TEA blockade effect on KCNQ1-4 channels in CHO cells, Hadley et al.[131] [145] found that KCNQ2 has the most robust sensitivity, KCNQ1 and KCNQ4 have intermediate sensitivity and KCNQ3 has little sensitivity, to external TEA. The differential sensitivity to TEA might be due to the tyrosine residue of the outer pore in KCNQ2 but lacking in other KCNQ channels that has been shown to be responsible for TEA binding in Kv channels [144][130]. In addition, internal TEA was found to inhibit KCNQ1 in the presence or absence of KCNE1 expressed in Xenopus oocytes [128][114]. Internal TEA binds to the intracellular pore and blocks the potassium current, which is the canonical pore occlusion mechanism. This blockage mechanism has been seen in other Kv channels [143,144][129][130]. However, KCNQ1/KCNE1 channels, compared to KCNQ1 channels, were shown to be more sensitive to internal TEA, suggesting KCNE1 helps determine the KCNQ1 pharmacological properties.
Chromanol 293B has been widely used as a specific KCNQ1/KCNE1 channel blocker and has been proposed as a potential class III antiarrhythmic agent. A class III antiarrhythmic agent acts by lengthening the repolarization phase of the cardiac action potential and causes a concomitant increase in the effective refractory period at slower heart rates [23][17]. Chromanol 293B was shown to block the guinea pig KCNQ1/KCNE1 channels expressed in Xenopus oocytes and the KCNQ1/KCNE1 current in guinea pig cardiomyocytes [22][16]. Chromanol 293B showed little effect on the IKr current which contributes to the depolarization of the cardiac action potential together with the KCNQ1/KCNE1 current in guinea pig cardiomyocytes [22][16]. In addition, it also exhibited no inhibitory effect on the cardiac hERG current expressed in Xenopus oocytes [22][16]. Bosch and his colleagues[15] [21] tested the effects of Chromanol 293B on the Kv, Nav and Cav currents and action potential in human and guinea pig cardiomyocytes. Chromanol 293B inhibits KCNQ1/KCNE1 but no other Kv (IK1 and Ito), Nav and Cav currents. It also prolongs the action potential duration in cardiomyocytes. These results together suggest that Chromanol 293B is a rather selective blocker for KCNQ1/KCNE1 channels. A previous study[117] [131] suggested that Chromanol 293B binds to KCNQ1 through electrostatic interaction with a potassium ion in the selectivity filter of the channel. HMR 1556, a chromanol derivative, has been shown to block the KCNQ1/KCNE1 channel with a higher binding affinity, compared with Chromanol 293B [133][119].
References
- H T Shih; Anatomy of the action potential in the heart.. Texas Heart Institute Journal 1994, 21, 30-41.
- Jeanne M. Nerbonne; Robert S. Kass; Molecular Physiology of Cardiac Repolarization. Physiological Reviews 2005, 85, 1205-1253, 10.1152/physrev.00002.2005.
- A. L. Hodgkin; A. F. Huxley; A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology 1952, 117, 500-544, 10.1113/jphysiol.1952.sp004764.
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996, 384, 80–83.
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996, 384, 78–80.
- Wang, K.; Terrenoire, C.; Sampson, K.J.; Iyer, V.; Osteen, J.D.; Lu, J.; Keller, G.; Kotton, D.N.; Kass, R.S. Biophysical properties of slow potassium channels in human embryonic stem cell derived cardiomyocytes implicate subunit stoichiometry. J. Physiol. 2011, 589, 6093–6104.
- Wang, M.; Kass, R.S. Stoichiometry of the slow Iks potassium channel in human embryonic stem cell-derived myocytes. Pediatr. Cardiol. 2012, 33, 938–942.
- Liin, S.I.; Barro-Soria, R.; Larsson, H.P. The KCNQ1 channel—Remarkable flexibility in gating allows for functional versatility. J. Physiol. 2015, 593, 2605–2615.
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908.
- Lu, Z.; Klem, A.M.; Ramu, Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 2002, 120, 663–676.
- Kalstrup, T.; Blunck, R. S4-S5 linker movement during activation and inactivation in voltage-gated K+ channels. Proc. Natl. Acad. Sci. USA 2018, 115, E6751–E6759.
- Liin, S.I.; Silvera Ejneby, M.; Barro-Soria, R.; Skarsfeldt, M.A.; Larsson, J.E.; Starck Harlin, F.; Parkkari, T.; Bentzen, B.H.; Schmitt, N.; Larsson, H.P.; et al. Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. USA 2015, 112, 5714–5719.
- Liin, S.I.; Larsson, J.E.; Barro-Soria, R.; Bentzen, B.H.; Larsson, H.P. Fatty acid analogue N-arachidonoyl taurine restores function of IKs channels with diverse long QT mutations. Elife 2016, 5, e20272.
- Bohannon, B.M.; de la Cruz, A.; Wu, X.; Jowais, J.J.; Perez, M.E.; Dykxhoorn, D.M.; Liin, S.I.; Larsson, H.P. Polyunsaturated fatty acid analogues differentially affect cardiac NaV, CaV, and KV channels through unique mechanisms. Elife 2020, 9, e51453.
- Bosch, R.F.; Gaspo, R.; Busch, A.E.; Lang, H.J.; Li, G.R.; Nattel, S. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc. Res. 1998, 38, 441–450.
- Busch, A.E.; Suessbrich, H.; Waldegger, S.; Sailer, E.; Greger, R.; Lang, H.; Lang, F.; Gibson, K.J.; Maylie, J.G. Inhibition of IKs in guinea pig cardiac myocytes and guinea pig IsK channels by the chromanol 293B. Pflüg. Arch. 1996, 432, 1094–1096.
- Selnick, H.G.; Liverton, N.J.; Baldwin, J.J.; Butcher, J.W.; Claremon, D.A.; Elliott, J.M.; Freidinger, R.M.; King, S.A.; Libby, B.E.; McIntyre, C.J.; et al. Class III antiarrhythmic activity in vivo by selective blockade of the slowly activating cardiac delayed rectifier potassium current IKs by (R)-2-(2,4-trifluoromethyl)-N-[2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)-2,3-dihydro-1H-benzo[e][1,4]diazepin-3-yl]acetamide. J. Med. Chem. 1997, 40, 3865–3868.
- Richard E. Klabunde; Cardiac electrophysiology: normal and ischemic ionic currents and the ECG. Advances in Physiology Education 2017, 41, 29-37, 10.1152/advan.00105.2016.
- Vincent G.M.; The long QT syndrome. Indian Pacing Electrophysiol J. 2002, 2, 127-142.
- Arpad Tosaki; ArrhythmoGenoPharmacoTherapy. Frontiers in Pharmacology 2020, 11, 616, 10.3389/fphar.2020.00616.
- Augustus O. Grant; Cardiac Ion Channels. Circulation: Arrhythmia and Electrophysiology 2009, 2, 185-194, 10.1161/circep.108.789081.
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT syndrome: From genetics to management. Circ. Arrhythm. Electrophysiol. 2012, 5, 868–877.
- Levine, E.; Rosero, S.Z.; Budzikowski, A.S.; Moss, A.J.; Zareba, W.; Daubert, J.P. Congenital long QT syndrome: Considerations for primary care physicians. Clevel. Clin. J. Med. 2008, 75, 591–600.
- Wu, J.; Sakaguchi, T.; Takenaka, K.; Toyoda, F.; Tsuji, K.; Matsuura, H.; Horie, M. A trafficking-deficient KCNQ1 mutation, T587M, causes a severe phenotype of long QT syndrome by interfering with intracellular hERG transport. J. Cardiol. 2019, 73, 343–350.
- Peng, G.; Barro-Soria, R.; Sampson, K.J.; Larsson, H.P.; Kass, R.S. Gating mechanisms underlying deactivation slowing by two KCNQ1 atrial fibrillation mutations. Sci. Rep. 2017, 7, 45911.
- Tamargo, J.; Caballero, R.; Gomez, R.; Valenzuela, C.; Delpon, E. Pharmacology of cardiac potassium channels. Cardiovasc. Res. 2004, 62, 9–33.
- Robbins, J. KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 2001, 90, 1–19. [Google Scholar] [CrossRef]
- Jespersen, T.; Grunnet, M.; Olesen, S.P. The KCNQ1 potassium channel: From gene to physiological function. Physiology 2005, 20, 408–416. [Google Scholar] [CrossRef]
- Cui, J. Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys. J. 2016, 110, 14–25.
- Abbott, G.W. Biology of the KCNQ1 potassium channel. New J. Sci. 2014, 2014, 237431.
- Tristani-Firouzi, M.; Sanguinetti, M.C. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 1998, 510 Pt 1, 37–45.
- Hou, P.; Eldstrom, J.; Shi, J.; Zhong, L.; McFarland, K.; Gao, Y.; Fedida, D.; Cui, J. Inactivation of KCNQ1 potassium channels reveals dynamic coupling between voltage sensing and pore opening. Nat. Commun. 2017, 8, 1730.
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e9.
- Sun, J.; MacKinnon, R. Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 2017, 169, 1042–1050.e9.
- Swartz, K.J. Sensing voltage across lipid membranes. Nature 2008, 456, 891–897.
- Zaydman, M.A.; Kasimova, M.A.; McFarland, K.; Beller, Z.; Hou, P.; Kinser, H.E.; Liang, H.; Zhang, G.; Shi, J.; Tarek, M.; et al. Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. Elife 2014, 3, e03606.
- Lacroix, J.J.; Pless, S.A.; Maragliano, L.; Campos, F.V.; Galpin, J.D.; Ahern, C.A.; Roux, B.; Bezanilla, F. Intermediate state trapping of a voltage sensor. J. Gen. Physiol. 2012, 140, 635–652.
- Delemotte, L.; Tarek, M.; Klein, M.L.; Amaral, C.; Treptow, W. Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 6109–6114.
- Taylor, K.C.; Kang, P.W.; Hou, P.; Yang, N.D.; Kuenze, G.; Smith, J.A.; Shi, J.; Huang, H.; White, K.M.; Peng, D.; et al. Structure and physiological function of the human KCNQ1 channel voltage sensor intermediate state. Elife 2020, 9, e53901.
- Wisedchaisri, G.; Tonggu, L.; McCord, E.; Gamal El-Din, T.M.; Wang, L.; Zheng, N.; Catterall, W.A. Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 2019, 178, 993–1003.e12.
- Yang, Y.; Sigworth, F.J. Single-channel properties of IKs potassium channels. J. Gen. Physiol. 1998, 112, 665–678.
- Werry, D.; Eldstrom, J.; Wang, Z.; Fedida, D. Single-channel basis for the slow activation of the repolarizing cardiac potassium current, IKs. Proc. Natl. Acad. Sci. USA 2013, 110, E996–E1005.
- Wang, Y.; Eldstrom, J.; Fedida, D. Gating and Regulation of KCNQ1 and KCNQ1 + KCNE1 Channel Complexes. Front. Physiol. 2020, 11, 504.
- Pusch, M.; Bertorello, L.; Conti, F. Gating and flickery block differentially affected by rubidium in homomeric KCNQ1 and heteromeric KCNQ1/KCNE1 potassium channels. Biophys. J. 2000, 78, 211–226.
- Seebohm, G.; Sanguinetti, M.C.; Pusch, M. Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. 2003, 552, 369–378.
- Schroeder, B.C.; Waldegger, S.; Fehr, S.; Bleich, M.; Warth, R.; Greger, R.; Jentsch, T.J. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 2000, 403, 196–199.
- Barro-Soria, R.; Ramentol, R.; Liin, S.I.; Perez, M.E.; Kass, R.S.; Larsson, H.P. KCNE1 and KCNE3 modulate KCNQ1 channels by affecting different gating transitions. Proc. Natl. Acad. Sci. USA 2017, 114, E7367–E7376.
- Leigh D. Plant; Dazhi Xiong; Hui Dai; Steve A.N. Goldstein; Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proceedings of the National Academy of Sciences 2014, 111, E1438-E1446, 10.1073/pnas.1323548111.
- Geoffrey W. Abbott; KCNE genetics and pharmacogenomics in cardiac arrhythmias: much ado about nothing?. Expert Review of Clinical Pharmacology 2013, 6, 49-60, 10.1586/ecp.12.76.
- Melman, Y.F.; Um, S.Y.; Krumerman, A.; Kagan, A.; McDonald, T.V. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron 2004, 42, 927–937.
- Melman, Y.F.; Domenech, A.; de la Luna, S.; McDonald, T.V. Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem. 2001, 276, 6439–6444.
- Panaghie, G.; Tai, K.K.; Abbott, G.W. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J. Physiol. 2006, 570, 455–467.
- Chung, D.Y.; Chan, P.J.; Bankston, J.R.; Yang, L.; Liu, G.; Marx, S.O.; Karlin, A.; Kass, R.S. Location of KCNE1 relative to KCNQ1 in the IKS potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. USA 2009, 106, 743–748.
- Nakajo, K.; Kubo, Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 2007, 130, 269–281.
- Xulin Xu; Min Jiang; Kai-Ling Hsu; Mei Zhang; Gea-Ny Tseng; KCNQ1 and KCNE1 in the IKs Channel Complex Make State-dependent Contacts in their Extracellular Domains. Journal of General Physiology 2008, 131, 589-603, 10.1085/jgp.200809976.
- Restier, L.; Cheng, L.; Sanguinetti, M.C. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J. Physiol. 2008, 586, 4179–4191.
- Chan, P.J.; Osteen, J.D.; Xiong, D.; Bohnen, M.S.; Doshi, D.; Sampson, K.J.; Marx, S.O.; Karlin, A.; Kass, R.S. Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J. Gen. Physiol. 2012, 139, 135–144.
- Cecile Terrenoire; Colleen E. Clancy; Joseph W. Cormier; Kevin J. Sampson; Robert S. Kass; Autonomic Control of Cardiac Action Potentials. Circulation Research 2005, 96, e25-e34, 10.1161/01.res.0000160555.58046.9a.
- Schwartz, P.J.; Priori, S.G.; Cerrone, M.; Spazzolini, C.; Odero, A.; Napolitano, C.; Bloise, R.; De Ferrari, G.M.; Klersy, C.; Moss, A.J.; et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 2004, 109, 1826–1833.
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk stratification in the long-QT syndrome. N. Engl. J. Med. 2003, 348, 1866–1874.
- Yazawa, K.; Kameyama, M. Mechanism of receptor-mediated modulation of the delayed outward potassium current in guinea-pig ventricular myocytes. J. Physiol. 1990, 421, 135–150.
- Volders, P.G.; Stengl, M.; van Opstal, J.M.; Gerlach, U.; Spatjens, R.L.; Beekman, J.D.; Sipido, K.R.; Vos, M.A. Probing the contribution of IKs to canine ventricular repolarization: Key role for beta-adrenergic receptor stimulation. Circulation 2003, 107, 2753–2760.
- Steven O. Marx; Junko Kurokawa; Steven Reiken; Howard Motoike; Jeanine D'armiento; Andrew R. Marks; Robert S. Kass; Requirement of a Macromolecular Signaling Complex for beta Adrenergic Receptor Modulation of the KCNQ1-KCNE1 Potassium Channel. Science 2002, 295, 496-499, 10.1126/science.1066843.
- Aflaki, M.; Qi, X.Y.; Xiao, L.; Ordog, B.; Tadevosyan, A.; Luo, X.; Maguy, A.; Shi, Y.; Tardif, J.C.; Nattel, S. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained beta-adrenergic activation in guinea pig hearts. Circ. Res. 2014, 114, 993–1003.
- Zhang, L.M.; Wang, Z.; Nattel, S. Effects of sustained beta-adrenergic stimulation on ionic currents of cultured adult guinea pig cardiomyocytes. Am. J. Physiol. Heart. Circ. Physiol. 2002, 282, H880–H889.
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195.
- Kruse, M.; Hille, B. The phosphoinositide sensitivity of the Kv channel family. Channels 2013, 7, 530–536.
- Hilgemann, D.W. On the physiological roles of PIP2 at cardiac Na+-Ca2+ exchangers and KATP channels: A long journey from membrane biophysics into cell biology. J. Physiol. 2007, 582, 903–909.
- Loussouarn, G.; Park, K.H.; Bellocq, C.; Baro, I.; Charpentier, F.; Escande, D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: A functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003, 22, 5412–5421.
- Li, Y.; Zaydman, M.A.; Wu, D.; Shi, J.; Guan, M.; Virgin-Downey, B.; Cui, J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. USA 2011, 108, 9095–9100.
- Li, Y.; Gamper, N.; Hilgemann, D.W.; Shapiro, M.S. Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 9825–9835.
- David Péroz; Nicolas Rodriguez; Frank Choveau; Isabelle Baró; Jean Mérot; Gildas Loussouarn; Kv7.1 (KCNQ1) properties and channelopathies. The Journal of Physiology 2008, 586, 1785-1789, 10.1113/jphysiol.2007.148254.
- Mark A. Zaydman; Jonathan R. Silva; Kelli Delaloye; Yang Li; Hongwu Liang; H. Peter Larsson; Jingyi Shi; Jianmin Cui; Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proceedings of the National Academy of Sciences 2013, 110, 13180-13185, 10.1073/pnas.1305167110.
- Robin Y. Kim; Stephan Alexander Pless; Harley T. Kurata; PIP2 mediates functional coupling and pharmacology of neuronal KCNQ channels.. Proceedings of the National Academy of Sciences 2017, 114, E9702-E9711, 10.1073/pnas.1705802114.
- Frank S. Choveau; Victor De La Rosa; Sonya M. Bierbower; Ciria C. Hernandez; Mark S. Shapiro; Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates KCNQ3 K+ channels by interacting with four cytoplasmic channel domains. Journal of Biological Chemistry 2018, 293, 19411-19428, 10.1074/jbc.ra118.005401.
- Yongfeng Liu; Xianjin Xu; Junyuan Gao; Moawiah M. Naffaa; Hongwu Liang; Jingyi Shi; Hong Zhan Wang; Nien-Du Yang; Panpan Hou; Wenshan Zhao; et al.Kelli McFarland WhiteWenjuan KongAlex DouAmy CuiGuohui ZhangIra S. CohenXiaoqin ZouJianmin Cui A PIP2 substitute mediates voltage sensor-pore coupling in KCNQ activation. Communications Biology 2020, 3, 1-12, 10.1038/s42003-020-1104-0.
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschutz, W.; Lipke, C.; Kostler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274.
- Li, Y.; Gao, J.; Lu, Z.; McFarland, K.; Shi, J.; Bock, K.; Cohen, I.S.; Cui, J. Intracellular ATP binding is required to activate the slowly activating K+ channel IKs. Proc. Natl. Acad. Sci. USA 2013, 110, 18922–18927.
- Burnstock, G.; Pelleg, A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2015, 11, 1–46.
- Hohl, C.M.; Hearse, D.J. Vascular and contractile responses to extracellular ATP: Studies in the isolated rat heart. Can. J. Cardiol. 1985, 1, 207–216.
- Zhang, B.X.; Desnoyer, R.W.; Bond, M. Extracellular Adenosine Triphosphate Triggers Arrhythmias and Elemental Redistribution in Electrically Stimulated Rat Cardiac Myocytes. Microsc. Microanal. 2001, 7, 48–55.
- Marie-Cécile Kienitz; Dilyana Vladimirova; Synergistic modulation of KCNQ1/KCNE1 K+ channels (IKs) by phosphatidylinositol 4,5-bisphosphate (PIP2) and [ATP]i. Cellular Signalling 2015, 27, 1457-1468, 10.1016/j.cellsig.2015.04.002.
- Naoya Oketani; Masafumi Kakei; Kotaro Ichinari; Midori Okamura; Akihiro Miyamura; Mitsuhiro Nakazaki; Seiki Ito; Chuwa Tei; Regulation of KATP channels by P2Ypurinoceptors coupled to PIP2 metabolism in guinea pig ventricular cells. American Journal of Physiology-Heart and Circulatory Physiology 2002, 282, H757-H765, 10.1152/ajpheart.00246.2001.
- Colin G. Nichols; Adenosine Triphosphate-Sensitive Potassium Currents in Heart Disease and Cardioprotection.. Cardiac Electrophysiology Clinics 2016, 8, 323-335, 10.1016/j.ccep.2016.01.005.
- Tosaki, A.; Hellegouarch, A. Adenosine triphosphate-sensitive potassium channel blocking agent ameliorates, but the opening agent aggravates, ischemia/reperfusion-induced injury. Heart function studies in nonfibrillating isolated hearts. J. Am. Coll. Cardiol. 1994, 23, 487–496.
- Miura, T.; Liu, Y.; Goto, M.; Tsuchida, A.; Miki, T.; Nakano, A.; Nishino, Y.; Ohnuma, Y.; Shimamoto, K. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2001, 37, 957–963.
- Billman, G.E. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol. Ther. 2008, 120, 54–70.
- Busch, A.E.; Herzer, T.; Wagner, C.A.; Schmidt, F.; Raber, G.; Waldegger, S.; Lang, F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol. Pharmacol. 1994, 46, 750–753.
- Abitbol, I.; Peretz, A.; Lerche, C.; Busch, A.E.; Attali, B. Stilbenes and fenamates rescue the loss of IKS channel function induced by an LQT5 mutation and other IsK mutants. EMBO J. 1999, 18, 4137–4148.
- Wang, Y.; Eldstrom, J.; Fedida, D. The IKs Ion Channel Activator Mefenamic Acid Requires KCNE1 and Modulates Channel Gating in a Subunit-Dependent Manner. Mol. Pharmacol. 2020, 97, 132–144.
- Hou, P.; Shi, J.; White, K.M.; Gao, Y.; Cui, J. ML277 specifically enhances the fully activated open state of KCNQ1 by modulating VSD-pore coupling. Elife 2019, 8, e48576.
- Mattmann, M.E.; Yu, H.; Lin, Z.; Xu, K.; Huang, X.; Long, S.; Wu, M.; McManus, O.B.; Engers, D.W.; Le, U.M.; et al. Identification of (R)-N-(4-(4-methoxyphenyl)thiazol-2-yl)-1-tosylpiperidine-2-carboxamide, ML277, as a novel, potent and selective Kv7.1 (KCNQ1) potassium channel activator. Bioorg. Med. Chem. Lett. 2012, 22, 5936–5941.
- Xu, Y.; Wang, Y.; Zhang, M.; Jiang, M.; Rosenhouse-Dantsker, A.; Wassenaar, T.; Tseng, G.N. Probing binding sites and mechanisms of action of an IKs activator by computations and experiments. Biophys. J. 2015, 108, 62–75.
- Yu, H.; Lin, Z.; Mattmann, M.E.; Zou, B.; Terrenoire, C.; Zhang, H.; Wu, M.; McManus, O.B.; Kass, R.S.; Lindsley, C.W.; et al. Dynamic subunit stoichiometry confers a progressive continuum of pharmacological sensitivity by KCNQ potassium channels. Proc. Natl. Acad. Sci. USA 2013, 110, 8732–8737.
- Mruk, K.; Kobertz, W.R. Discovery of a novel activator of KCNQ1-KCNE1 K channel complexes. PLoS ONE 2009, 4, e4236.
- Xiong, Q.; Sun, H.; Li, M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat. Chem. Biol. 2007, 3, 287–296.
- ANGELE De Silva; Rían W. Manville; Geoffrey W. Abbott; Deconstruction of an African folk medicine uncovers a novel molecular strategy for therapeutic potassium channel activation. Science Advances 2018, 4, eaav0824, 10.1126/sciadv.aav0824.
- Main, M.J.; Cryan, J.E.; Dupere, J.R.; Cox, B.; Clare, J.J.; Burbidge, S.A. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 2000, 58, 253–262.
- Tatulian, L.; Delmas, P.; Abogadie, F.C.; Brown, D.A. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J. Neurosci. 2001, 21, 5535–5545.
- Manville, R.W.; Papanikolaou, M.; Abbott, G.W. Direct neurotransmitter activation of voltage-gated potassium channels. Nat. Commun. 2018, 9, 1847.
- Kim, R.Y.; Yau, M.C.; Galpin, J.D.; Seebohm, G.; Ahern, C.A.; Pless, S.A.; Kurata, H.T. Atomic basis for therapeutic activation of neuronal potassium channels. Nat. Commun. 2015, 6, 8116.
- Briana M. Bohannon; Xiaoan Wu; Xiongyu Wu; Marta E. Perez; Sara I. Liin; H. Peter Larsson; Polyunsaturated fatty acids produce a range of activators for heterogeneous IKs channel dysfunction. Journal of General Physiology 2019, 152, e201912396, 10.1085/jgp.201912396.
- Takeshi Aiba; Wataru Shimizu; Masashi Inagaki; Takashi Noda; Shunichiro Miyoshi; Wei-Guang Ding; Dimitar P. Zankov; Futoshi Toyoda; Hiroshi Matsuura; Minoru Horie; et al.Kenji Sunagawa Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. Journal of the American College of Cardiology 2005, 45, 300-307, 10.1016/j.jacc.2004.09.069.
- Mark Alexander Skarsfeldt; Sara I. Liin; Hans P. Larsson; Bo H. Bentzen; Polyunsaturated fatty acid‐derived I Ks channel activators shorten the QT interval ex‐vivo and in‐vivo. Acta Physiologica 2020, 229, e13471, 10.1111/apha.13471.
- Paula L. Hedley; Poul Jørgensen; Sarah Schlamowitz; Romilda Wangari; Johanna Moolman-Smook; Paul A. Brink; Jørgen K. Kanters; Valerie A. Corfield; Michael Christiansen; The genetic basis of long QT and short QT syndromes: A mutation update. Human Mutation 2009, 30, 1486-1511, 10.1002/humu.21106.
- Benatti, P.; Peluso, G.; Nicolai, R.; Calvani, M. Polyunsaturated fatty acids: Biochemical, nutritional and epigenetic properties. J. Am. Coll. Nutr. 2004, 23, 281–302.
- Wiktorowska-Owczarek, A.; Berezinska, M.; Nowak, J.Z. PUFAs: Structures, Metabolism and Functions. Adv. Clin. Exp. Med. 2015, 24, 931–941.
- Liin, S.I.; Yazdi, S.; Ramentol, R.; Barro-Soria, R.; Larsson, H.P. Mechanisms Underlying the Dual Effect of Polyunsaturated Fatty Acid Analogs on Kv7.1. Cell Rep. 2018, 24, 2908–2918.
- Borjesson, S.I.; Hammarstrom, S.; Elinder, F. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 2008, 95, 2242–2253.
- Briana M. Bohannon; Marta E. Perez; Sara I. Liin; H. Peter Larsson; ω-6 and ω-9 polyunsaturated fatty acids with double bonds near the carboxyl head have the highest affinity and largest effects on the cardiac I K s potassium channel. Acta Physiologica 2018, 225, e13186-e13186, 10.1111/apha.13186.
- Y. F. Xiao; J. X. Kang; J. P. Morgan; A. Leaf; Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes.. Proceedings of the National Academy of Sciences 1995, 92, 11000-11004, 10.1073/pnas.92.24.11000.
- J. X. Kang; Y. F. Xiao; A. Leaf; Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes.. Proceedings of the National Academy of Sciences 1995, 92, 3997-4001, 10.1073/pnas.92.9.3997.
- Yong-Fu Xiao; Ana Maria Gomez; James P. Morgan; W. J. Lederer; Alexander Leaf; Suppression of voltage-gated L-type Ca2+ currents by polyunsaturated fatty acids in adult and neonatal rat ventricular myocytes. Proceedings of the National Academy of Sciences 1997, 94, 4182-4187, 10.1073/pnas.94.8.4182.
- Sesti, F.; Tai, K.K.; Goldstein, S.A. MinK endows the IKs potassium channel pore with sensitivity to internal tetraethylammonium. Biophys. J. 2000, 79, 1369–1378.
- Tai, K.K.; Goldstein, S.A. The conduction pore of a cardiac potassium channel. Nature 1998, 391, 605–608.
- Kurokawa, J.; Motoike, H.K.; Kass, R.S. TEA+-sensitive KCNQ1 constructs reveal pore-independent access to KCNE1 in assembled IKs channels. J. Gen. Physiol. 2001, 117, 43–52.
- Christian Lerche; Iva Bruhova; Holger Lerche; Klaus Steinmeyer; Aguan D. Wei; Nathalie Strutz-Seebohm; Florian Lang; Andreas E. Busch; Boris S. Zhorov; Guiscard Seebohm; et al. Chromanol 293B Binding in KCNQ1 (Kv7.1) Channels Involves Electrostatic Interactions with a Potassium Ion in the Selectivity Filter. Molecular Pharmacology 2007, 71, 1503-1511, 10.1124/mol.106.031682.
- Guiscard Seebohm; Jun Chen; Nathalie Strutz; Chris Culberson; Christian Lerche; Michael C. Sanguinetti; Molecular Determinants of KCNQ1 Channel Block by a Benzodiazepine. Molecular Pharmacology 2003, 64, 70-77, 10.1124/mol.64.1.70.
- Thomas, G.P.; Gerlach, U.; Antzelevitch, C. HMR 1556, a potent and selective blocker of slowly activating delayed rectifier potassium current. J. Cardiovasc. Pharmacol. 2003, 41, 140–147.
- Gogelein, H.; Bruggemann, A.; Gerlach, U.; Brendel, J.; Busch, A.E. Inhibition of IKs channels by HMR 1556. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 480–488.
- Kang, C.; Badiceanu, A.; Brennan, J.A.; Gloschat, C.; Qiao, Y.; Trayanova, N.A.; Efimov, I.R. β-adrenergic stimulation augments transmural dispersion of repolarization via modulation of delayed rectifier currents IKs and IKr in the human ventricle. Sci. Rep. 2017, 7, 15922.
- Bjork, S.; Ojala, E.A.; Nordstrom, T.; Ahola, A.; Liljestrom, M.; Hyttinen, J.; Kankuri, E.; Mervaala, E. Evaluation of Optogenetic Electrophysiology Tools in Human Stem Cell-Derived Cardiomyocytes. Front. Physiol. 2017, 8, 884.
- Heun Soh; Anastasios V. Tzingounis; The Specific Slow Afterhyperpolarization Inhibitor UCL2077 Is a Subtype-Selective Blocker of the Epilepsy Associated KCNQ Channels. Molecular Pharmacology 2010, 78, 1088-1095, 10.1124/mol.110.066100.
- H S Wang; B S Brown; D McKinnon; I S Cohen; Molecular basis for differential sensitivity of KCNQ and I(Ks) channels to the cognitive enhancer XE991.. Molecular Pharmacology 2000, 57, 1218–1223.
- Kathya Villatoro-Gómez; David O. Pacheco-Rojas; Eloy G. Moreno-Galindo; Ricardo A. Navarro-Polanco; Martin Tristani-Firouzi; Dimitris Gazgalis; Meng Cui; José A. Sánchez-Chapula; Tania Ferrer; Molecular determinants of Kv7.1/KCNE1 channel inhibition by amitriptyline. Biochemical Pharmacology 2018, 152, 264-271, 10.1016/j.bcp.2018.03.016.
- Meikui Wu; Makoto Takemoto; Huan Luo; Jianjun Xu; Mei-Hong Lu; Masaki Kameyama; Toru Takumi; Wen-Jie Song; A novel role of the antitumor agent tricyclodecan-9-yl-xanthogenate as an open channel blocker of KCNQ1/KCNE1. European Journal of Pharmacology 2018, 824, 99-107, 10.1016/j.ejphar.2018.02.013.
- Minghua Wu; Yutaro Obara; Ikuo Norota; Yoshinobu Nagasawa; Kuniaki Ishii; Insulin suppresses IKs (KCNQ1/KCNE1) currents, which require β-subunit KCNE1. Pflügers Archiv 2013, 466, 937-946, 10.1007/s00424-013-1352-7.
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77.
- MacKinnon, R.; Yellen, G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science 1990, 250, 276–279.
- Heginbotham, L.; MacKinnon, R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron 1992, 8, 483–491.
- J K Hadley; M Noda; A A Selyanko; I C Wood; F C Abogadie; D A Brown; Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. British Journal of Pharmacology 2000, 129, 413-415, 10.1038/sj.bjp.0703086.