KLF11 (Krüppel-like factor 11) belongs to the family of Sp1/Krüppel-like zinc finger transcription factors that play important roles in a variety of cell types and tissues. In carcinogenesis, KLF11 can show diverse effects. Its function as a tumor suppressor gene can be suppressed by phosphorylation of its binding domains via oncogenic pathways. However, KLF 11 itself can also show tumor-promoting effects and seems to have a crucial role in the epithelial-mesenchymal transition (EMT) process.
- KLF11
- TGF-β
- TIEG
- Cell cycle
- Apoptosis
- Cell growth
- Cancer
Given the described role of KLF11 in growth regulation, it is not surprising that KLF11 has also been implicated in the development of tumors. In carcinogenesis, the TGF-β signaling pathway plays a dual role characterized by tumor suppression at early tumor stages and enhanced tumor progression at the late stages of the disease [43,59,60[1][2][3]]. TGF-β mediates tumor suppression via Smad-dependent signaling, which then regulates the transcription of cell-cycle-associated genes like p15 and p21 [44,61[4][5]]. But Smad7 also exerts a negative feedback loop by binding to the activated TβR-I, blocking it and preventing phosphorylation [62,63[6][7]]. Disturbances in the Smad-dependent signaling have recently been shown in human cancers (including pancreatic cancer), and are associated with the ability of tumor cells to escape from the TGF-β–induced growth inhibition [64,65[8][9]]. TGF-β may also signal through Smad-independent signaling cascades (e.g., Rho-like guanosine triphosphatases, p38, mitogen-activated protein kinase [MAPK], phosphatidylinositol-3-kinase or c-Jun-N-terminal kinase) and induce an epithelial-to-mesenchymal transition (EMT)—which is a key process in the formation of cancer metastasis—of tumor cells, leading to enhanced tumor cell migration and invasion [66–70[10][11][12][13][14]].
KLF11, as discussed above, is an early response transcription factor that potentiates the TGF-β induced growth inhibition in normal epithelial cells by terminating the inhibitory Smad7 loop. In pancreatic cancer cells with oncogenic Ras mutations, this function of KLF11 is inhibited by the oncogenic Erk/MAPK: Erk/MAPK phosphorylates KLF11, which leads to the disruption of the KLF11–mSin3a interaction [46[15]]. This inhibits the binding of the KLF11–Smads complex to the TIE element and leads to a reduced TGF-β-induced c-myc repression and to a reduction of the anti-proliferative effects of TGF-β [40[16]]. Another study showsd that KLF11 is inhibited in CHO cells by the epidermal growth factor (EGF)–Ras–MEK1–ERK2 signaling pathway. Like in the Erk/MAPK pathway, phosphorylation of four serine/threonine sites adjacent to the SID leads to the disruption of the SID–mSin3A interaction [24[17]]. In KRAS oncogenic mutant cancer cells, KLF11 inhibits BrdU incorporation, increases apoptosis, and inhibits the KRAS-mediated foci and agar colony formation. In vivo, KLF11 partly inhibits the growth of pancreatic tumor cells of KRAS mutant xenografts, by inducing cell cycle arrest at the S phase via downregulation of cyclin A2 [11[18]]. Therefore, KLF11 might participate in the functional switch of TGF-β from a tumor suppressor to a tumor promoter. Figure 1 gives an overview of the different roles of the KLF11-mediated TGF-β–TGF-receptor–Smad signaling pathway in normal cells and tumors.
In addition to the inhibition of KLF11 by SID phosphorylation via Erk/MAPK, Buttar et al. [25[19]] suggested an additional model of KLF11-mediated tumor suppression and its antagonism by an oncogenic pathway. KLF11 binds to the GC-rich consensus sequences in the promoter region of cPLA2α, the key rate-limiting enzyme of the oncogenic PGE2 cascade. Following binding, KLF11 represses the cPLA2α promoter by recruiting the chromatin-remodeling complex Sin3a-HDAC to the promotor. In this way, KLF11 behaves as a tumor suppressor gene by repression of the cPLA2α–PGE2 pathway. This mechanism was shown in Barrett’s epithelial cells. EGFR-AKT signaling, which is upregulated in a subset of patients during carcinogenesis in Barrett’s esophagus cancer, leads to the phosphorylation of threonine at position 56 in the R1 domain (SID) of KLF11. This phosphorylation inhibits the KLF11 binding, and the repression of the cPLA2α promoter and the tumor-suppressing effects of KLF11 are inhibited. Probably EGFR can use two different intracellular pathways (ERK2 versus AKT) to inactivate KLF11 via phosphorylation. These phosphorylation events expand our biochemical knowledge about KLF11 to the post-translational effects.
On the contrary, direct tumor-promoting effects of KLF11 have also been described. In hepatocellular carcinoma, KLF11 had a significant influence on proliferation and apoptosis. It also promoted local invasion and distant migration by suppressing the Smad7 transcription via binding to the Smad7 promoter or by directly upregulating the Smad2/3 expression [71,72[20][21]]. Ji Q et al. demonstrated that the relative Twist1 promoter region activity increased gradually with increasing KLF11 levels in the plasma [73[22]]. Therefore, they speculated that KLF11 might regulate gastric cancer migration and invasion by increasing the Twist1 expression, which is essential for EMT [73[22]].
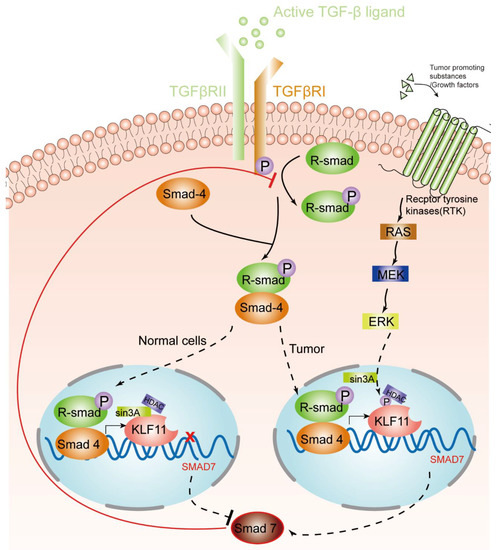
Figure 1. KLF11-mediated modulation of the TGF-β signaling pathway in normal cells and tumors. An activated TGF-β ligand binds to the type 2 domain of the TGF-β receptor, which then recruits and phosphorylates a type 1 receptor. The type 1 receptor then recruits and phosphorylates a receptor-regulated Smad (R-smad). The R-smad then binds to the common smad, Smad 4, and forms a heterodimeric complex. The Smad complex translocates to the nucleus to induce the expression of KLF11. In normal cells, the KLF11-Sin3A-HDAC complex binds to the promoters of Smad7 and represses its expression, which acts as a negative feedback loop of the TGF-β–Smads signaling pathway. However, in some tumors, RAS was activated by tumor-promoting substances/growth factors. Ras phosphates KLF11 by the RAS–MEK–ERK pathway, which leads to the disruption of the KLF11–mSin3a interaction, resulting in the termination of the inhibitory KLF11-mediated Smad7 loop.
KLF11-methylation-dependent inactivation and downregulation occurs in several malignancies, including leukemia, myeloproliferative disorders, esophageal adenocarcinoma, pancreatic cancer, germ cell tumors, ovarian cancer, and head and neck cancer, supporting its candidacy as an actual tumor suppressor gene in humans [25,37,74–78[19][23][24][25][26][27][28]]. KLF11 is involved in the progression of a wide variety of cancers, such as ovarian cancer and pancreatic cancer [40,78[16][28]]. In breast cancer, the KLF11 promotor is also hypermethylated, and the hypermethylation is associated with low expression of KLF11. KLF11 hypermethylation might be associated with higher rates of metastases [79[29]]. Similar results were found in uterine fibroids and in myelodysplastic syndrome [74,80[24][30]], suggesting that DNA methylation to regulate KLF11 expression might be a key event that directly contributes to tumorigenesis.
It was recently reported that the microRNA miR-30d increased the survival of BT474 and MDA-MB-231 breast cancer cells. It was shown that it inhibited apoptosis and increased Bcl-2 expression, while it reduced the Bax protein levels. This influence of miR-30d on breast cancer cell growth, metastasis, and EMT is dependent on a low level of KLF11 and on a high level of pSTAT3. KLF11 is a direct target of miR-30d, and KLF11 and pSTAT3 expression are regulated by miR-30d [81[31]].
MiR-30 also reduced the profibrogenic TGF-β signaling in hepatic stellate cells by suppressing the KLF11 expression and thus enhancing the negative feedback loop of TGF-β signaling imposed by Smad7 [82[32]]. LincRNA-p21 reduced the availability of miR-30 [83[33]]. Besides miR-30d, overexpression of miR-10b in hepatocellular carcinoma (HCC) promoted HCC cell migration and invasion. MiR-10b downregulated KLF4, which is the inhibitory transcriptional factor of KLF11. In this way, KLF11 was upregulated, which promoted HCC EMT [72[21]].
All these results suggest that KLF11 plays a crucial role during tumorigenesis and development. (Table 31.).
Table 31.
KLF11 in cancers.
Role in Cancer | Demonstrated Functional Effects | Cancers/Cancer Cell Type | Date | Ref. | ||||||||||
KLF11 could be induced in non-small cell lung cancer by radiohyperthermia and might mediate the effects of radiohyperthermia. | KLF11 induced apoptosis and inhibited cell proliferation by elevating intracellular reactive oxygen species. KLF11 knockdown reduced the effects of radiohyperthermia. | Human non-small-cell lung cancer | 2019 | [84] | [34]] |
|||||||||
KLF11 mediates the tumor-promoting effects of miRNA-30d in breast cancer. | MiRNA-30d increases breast cancer cell survival, inhibits apoptosis, promotes migration and invasion, and mediates the epithelial–mesenchymal transition (EMT) phenotype. MiRNA-30d exerts these effects by targeting KLF11 and activating the STAT3 pathway. | Breast cancer | 2018 | [81] | [31]] |
|||||||||
KLF11-methylation might be a biomarker for breast cancer diagnosis and prognosis. | The median methylation levels of KLF11 were ≥30% higher than in normal samples. KLF11 methylation might also be associated with a higher risk of metastasis. | Breast cancer | 2012 | [79] | [29]] |
|||||||||
KLF11 expression is reduced in ovarian cancer. | KLF11 promoter DNA methylation results in downregulated KLF11 expression accompanied by reduced Smad2, Smad3, and Smad7 expression | Human ovarian cancer | 2015 | [78] | [28]] |
|||||||||
KLF11 is upregulated in gastric cancer an increases gastric cancer cell migration and invasion. | KLF11 increases the Twist-1 expression in gastric cancer cells. The Twist-1 increase is inhibited when KLF11 is silenced. | Human gastric cancer | 2019 | [73] | [22]] |
|||||||||
KLF11 inhibits prostaglandin E2 (PGE2) synthesis. | KLF11 represses the promotor of the PGE2-synthesizing enzyme cytosolic phospholipase A2∝ by binding and by recruiting the Sin3-histone deacetylase chromatin remodeling complex to the promotor. | Esophageal cancer (Barretts’ esophageal cells) | 2010 | [25] | [19]] |
|||||||||
KLF11 mediates the-promoting effects of miRNA-10b on EMT development in hepatocellular carcinoma. | MiRNA-10b binds to the 3’UTR and downregulates KLF4, which is an inhibitory transcriptional factor of KLF11. Thereby, KLF11 is upregulated and reduces the expression ofSmad7. This upregulates Smad3, which promotes EMT development. | Human hepatocellular carcinoma | 2018 | [72] | [21]] |
|||||||||
KLF11 increases the monoamine oxidase (MAO) B expression. | KLF11 increases MAO B at the promotor activity, mRNA, protein, and catalytic activity levels. | Neuroblastoma and liver carcinoma cells | 2004 | [85] | [35]] |
|||||||||
KLF11 uses the epigenetic regulator heterochromatin protein 1 (HP1) to mediate tumor suppression. | KLF11 recruits HP1 and its histone methyltransferase to promotors of cancer genes to limit the KLF11-mediated gene activation. The impairment of this recruitment impairs tumor suppression. | Pancreatic cancer cells | 2012 | [86] | [36]] |
|||||||||
KLF11 mediates growth inhibition; the mechanism is disrupted in pancreatic cancer. | In pancreatic cancer cells, the KLF11–Smad3 complex formation is disrupted and the KLF11–Smad3 binding to the TGF-β-inhibitory element of the c-myc-promotor is inhibited. Thereby, the growth inhibitory effect of c-myc-silencing is impaired. | Pancreatic cancer cells | 2006 | [40] | [16]] |
|||||||||
The KLF11-induced potentiating of the TGF-β-signaling by the termination of the inhibitory Smad7-loop is inhibited in pancreatic cancer. | In pancreatic cancer cells, an Erk/mitogen-activated protein kinase phosphorylates KLF11, which leads to a disruption of the KLF11–mSin3a interaction. The KLF11–mSin3a repression of the Smad7 promotor is reduced, and therefore, Smad7 expression is elevated and Smad7 exerts its negative feedback loop. | Pancreatic cancer cells | 2004 | [46] | [15]] |
|||||||||
KLF11 is a tumor-suppressor gene inactivated in myelodysplastic syndromes (MDS). | KLF11 is hypermethylated in 15 % of MDS cases, which is associated with a high International Prognostic Scoring System score. | Human myelogenous leukemia cells | 2010 | [74] | [24]] |
References
- Rik Derynck; Ying E. Zhang; Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577-584, 10.1038/nature02006.
- Fan, Y.; Konermann, C.; Aulmann, S.; Bermejo, J.L.; Brugger, M.; Diederichs, S.; Rom, J.; Weichenhan, D.; Claus, R.; Rehli, M.; et al.et al. Kruppel-like factor-11, a transcription factor involved in diabetes mellitus, suppresses endothelial cell activation via the nuclear factor-kappaB signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2981–2988.
- Grunewald, M.; Johnson, S.; Lu, D.; Wang, Z.; Lomberk, G.; Albert, P.R.; Stockmeier, C.A.; Meyer, J.H.; Urrutia, R.; Miczek, K.A.; et al.et al. Mechanistic Role for a Novel Glucocorticoid-KLF11 (TIEG2) Protein Pathway in Stress-induced Monoamine Oxidase A Expression*. Journal of Biological Chemistry 2012, 287, 24195-24206, 10.1074/jbc.M112.373936.
- Joan Massagué; David Wotton; Transcriptional control by the TGF-beta/Smad signaling system. The EMBO Journal 2000, 19, 1745-1754, 10.1093/emboj/19.8.1745.
- Ken-Ichi Yamamoto; Masakiyo Sakaguchi; Reinhold J. Medina; Aya Niida; Yoshihiko Sakaguchi; Masahiro Miyazaki; Ken Kataoka; Nam-Ho Huh; Transcriptional regulation of a brown adipocyte-specific gene, UCP1, by KLF11 and KLF15. Biochemical and Biophysical Research Communications 2010, 400, 175-180, 10.1016/j.bbrc.2010.08.039.
- X. Niu; N. Perakakis; K. Laubner; C. Limbert; T. Stahl; M. D. Brendel; R. G. Bretzel; J. Seufert; G. Päth; Human Krüppel-like factor 11 inhibits human proinsulin promoter activity in pancreatic beta cells. Diabetologia 2007, 50, 2031-2031, 10.1007/s00125-007-0770-5.
- Neve, B.; Fernandez-Zapico, M.E.; Ashkenazi-Katalan, V.; Dina, C.; Hamid, Y.H.; Joly, E.; Vaillant, E.; Benmezroua, Y.; Durand, E.; Bakaher, N.; et al.et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proceedings of the National Academy of Sciences 2005, 102, 4807-4812, 10.1073/pnas.0409177102.
- Rosemary J. Akhurst; Rik Derynck; TGF-beta signaling in cancer--a double-edged sword. Trends in Cell Biology 2001, 11, S44–S51.
- Xin-Lin Chen; Zhuo-Qun Chen; Shui-Lian Zhu; Tian-Wen Liu; Yi Wen; Yi-Sheng Su; Xu-Jie Xi; Yue Hu; Lei Lian; Feng-Bin Liu; et al. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: a meta-analysis. BMC Cancer 2017, 17, 240, 10.1186/s12885-017-3215-7.
- Yigong Shi; Joan Massagué; Mechanisms of TGF-beta signaling from cell membrane to the nucleus.. Cell 2003, 113, , null.
- H Hayashi; S Abdollah; Y Qiu; J Cai; Y Y Xu; B W Grinnell; M A Richardson; James N. Topper; M A Gimbrone; J L Wrana; D Falb; The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling.. Cell 1997, 89, , null.
- Atsuhito Nakao; Mozhgan Afrakhte; Anita Morn; Takuya Nakayama; Jan L. Christian; Rainer Heuchel; Susumu Ltoh; Masahiro Kawabata; Nils-Erik Heldin; Carl-Henrik Heldin; Peter Ten Dijke; A Morén; Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631-635, 10.1038/39369.
- Veronica V. Rossato; Daner A. Silveira; Shantanu Gupta; José Carlos M. Mombach; Towards the contribution of the p38MAPK pathway to the dual role of TGFβ in cancer: A boolean model approach. Computers in Biology and Medicine 2019, 104, 235-240, 10.1016/j.compbiomed.2018.11.025.
- Nancy Dumont; Carlos L. Arteaga; Targeting the TGF beta signaling network in human neoplasia.. Cancer Cell 2003, 3, , null.
- Johnsen, S.A.; Subramaniam, M.; Janknecht, R.; Spelsberg, T.C.; TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene 2002, 21, 5783–5790.
- Buck, A.; Buchholz, M.; Wagner, M.; Adler, G.; Gress, T.; Ellenrieder, V.; The tumor suppressor KLF11 mediates a novel mechanism in transforming growth factor beta-induced growth inhibition that is inactivated in pancreatic cancer. Mol. Cancer Res. 2006, 4, 861–872.
- Volker Ellenrieder; Jin‐San Zhang; Joanna Kaczynski; Raul Urrutia; Signaling disrupts mSin3A binding to the Mad1-like Sin3-interacting domain of TIEG2, an Sp1-like repressor. The EMBO Journal 2002, 21, 2451-2460, 10.1093/emboj/21.10.2451.
- Martin E. Fernandez‑Zapico; Gwen A. Lomberk; Shoichiro Tsuji; Cathrine J. Demars; Michael R. Bardsley; Yi‑Hui Lin; Luciana L. Almada; Jing‑Jing Han; Debabrata Mukhopadhyay; Tamas Ordog; et al.Navtej S. ButtarRaul Urrutia A functional family-wide screening of SP/KLF proteins identifies a subset of suppressors of KRAS-mediated cell growth. Biochemical Journal 2011, 435, 529-537, 10.1042/bj20100773.
- Navtej Buttar; Cathrine J. Demars; Gwen Lomberk; Sumera Rizvi; Juliana Bonilla-Velez; Shalini Achra; Shahrooz Rashtak; Kenneth K. Wang; Martin E. Fernandez-Zapico; Raul Urrutia; et al. Distinct Role of Kruppel-like Factor 11 in the Regulation of Prostaglandin E2 Biosynthesis*. Journal of Biological Chemistry 2010, 285, 11433-11444, 10.1074/jbc.M109.077065.
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E.; TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759.
- Martin E. Fernandez-Zapico; Ann Mladek; Volker Ellenrieder; Emma Folch-Puy; Laurence Miller; Raul Urrutia; An mSin3A interaction domain links the transcriptional activity of KLF11 with its role in growth regulation. The EMBO Journal 2003, 22, 4748-4758, 10.1093/emboj/cdg470.
- W Cui; D J Fowlis; S Bryson; E Duffie; H Ireland; A Balmain; Rosemary J. Akhurst; TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice.. Cell 1996, 86, , null.
- V Ellenrieder; S F Hendler; W Boeck; T Seufferlein; Andre Menke; C Ruhland; G Adler; Thomas M Gress; Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation.. Cancer Research 2001, 61, , null.
- Jung, C.J.; Liengar, S.; lyengar, S.; Blahbik, K.R.; Jiang, J.X.; Tahimic, C.; Torok, N.J.; de vere White, R. W.; Farnham, P.J.; Zern, M. Human ESC self-renewal promoting microRNAs induce epithelial-mesenchymal transition in hepatocytes by controlling the PTEN and TGFbeta tumor suppressor signaling pathways. Mol. Cancer Res. 2012, 10, 979–991. [Google Scholar] [CrossRef]
- Gulibaha Hujie; Sheng-Hua Zhou; Hua Zhang; Jie Qu; Xiao-Wei Xiong; Outikuer Hujie; Cheng-Gong Liao; Shun-E Yang; MicroRNA-10b regulates epithelial-mesenchymal transition by modulating KLF4/KLF11/Smads in hepatocellular carcinoma.. Cancer Cell International 2018, 18, 10, 10.1186/s12935-018-0508-0.
- Q. Ji; Y. Li; Q. Zhao; L. Q. Fan; B. B. Tan; Z. D. Zhang; X. F. Zhao; Y. Liu; D. Wang; N. Jia; KLF11 promotes gastric cancer invasion and migration by increasing Twist1 expression. Neoplasma 2019, 66, 92-100, 10.4149/neo_2018_180325n201.
- Anna Potapova; Britta Hasemeier; Daniel Römermann; Kathleen Metzig; Gudrun Göhring; Brigitte Schlegelberger; Florian Länger; Hans Kreipe; Ulrich Lehmann; Epigenetic inactivation of tumour suppressor geneKLF11in myelodysplastic syndromes. European Journal of Haematology 2010, 84, 298-303, 10.1111/j.1600-0609.2009.01389.x.
- Hendrik Wermann; Hans Stoop; Ad Jm Gillis; Friedemann Honecker; Ruud Jhlm Van Gurp; Ole Ammerpohl; Julia Richter; J Wolter Oosterhuis; Carsten Bokemeyer; Leendert H. J. Looijenga; et al. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. The Journal of Pathology 2010, 221, 433–442, 10.1002/path.2725.
- Claudia Gebhard; Lucia Schwarzfischer; Thu-Hang Pham; Elmar Schilling; Maja Klug; R Andreesen; Michael Rehli; Genome-Wide Profiling of CpG Methylation Identifies Novel Targets of Aberrant Hypermethylation in Myeloid Leukemia. Cancer Research 2006, 66, 6118-6128, 10.1158/0008-5472.can-06-0376.
- Alejandro Schaffer; Belbin Tj; Singh B; Barber I; Socci N; Wenig B; Smith R; Prystowsky Mb; Childs G; Faculty of 1000 evaluation for Molecular classification of head and neck squamous cell carcinoma using cDNA microarrays.. F1000 - Post-publication peer review of the biomedical literature 2002, 62, 1184–1190, 10.3410/f.1007599.96155.
- Guan Wang; Xianfeng Li; Weiping Tian; Yan Wang; Dandan Wu; Zhongsheng Sun; Enfeng Zhao; Promoter DNA methylation is associated withKLF11expression in epithelial ovarian cancer. Genes, Chromosomes and Cancer 2015, 54, 453-462, 10.1002/gcc.22257.
- Marta Faryna; Carolin Konermann; Sebastian Aulmann; Justo Lorenzo Bermejo; Markus Brugger; Sven Diederichs; Joachim Rom; Dieter Weichenhan; Rainer Claus; M. Rehli; et al.Peter SchirmacherHans-Peter SinnChristoph PlassClarissa Gerhauser Genome‐wide methylation screen in low‐grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. The FASEB Journal 2012, 26, 4937-4950, 10.1096/fj.12-209502.
- Antonia Navarro; Ping Yin; Diana Monsivais; Simon M. Lin; Pan Du; Jian-Jun Wei; Serdar E Bulun; Genome-Wide DNA Methylation Indicates Silencing of Tumor Suppressor Genes in Uterine Leiomyoma. PLOS ONE 2012, 7, e33284, 10.1371/journal.pone.0033284.
- Mingli Han; Yimeng Wang; Guangcheng Guo; Lin Li; Dongwei Dou; Xin Ge; Pengwei Lv; Fang Wang; Yuanting Gu; microRNA-30d mediated breast cancer invasion, migration, and EMT by targeting KLF11 and activating STAT3 pathway. Journal of Cellular Biochemistry 2018, 119, 8138-8145, 10.1002/jcb.26767.