Alzheimer’s disease (AD) is characterized by the formation of intracellular aggregate composed of heavily phosphorylated tau protein and extracellular deposit of amyloid-β (Aβ) plaques derived from proteolysis cleavage of amyloid precursor protein (APP). Autophagy refers to the lysosomal-mediated degradation of cytoplasmic constituents, which plays a critical role in maintaining cellular homeostasis. Importantly, recent studies reported that dysregulation of autophagy is associated in the pathogenesis of AD, and therefore, autophagy modulation has gained attention as a promising approach to treat AD pathogenesis. In AD, both the maturation of autolysosomes and its retrograde transports have been obstructed, which causes the accumulation of autophagic vacuoles and eventually leads to degenerating and dystrophic neurites function. However, the mechanism of autophagy modulation in APP processing and its pathogenesis have not yet been fully elucidated in AD. In the early stage of AD, APP processing and Aβ accumulation-mediated autophagy facilitate the removal of toxic protein aggregates via mTOR-dependent and -independent pathways. In addition, a number of autophagy-related genes (Atg) and APP are thought to influence the development of AD, providing a bidirectional link between autophagy and AD pathology.
- Alzheimer’s Disease
- Autophagy
- Amyloid precursor protein
- Alzheimer’s Disease,Autophagy,Amyloid precursor protein
1. Introduction
Alzheimer’s disease (AD) is the most irreversible and progressive brain disorder of neurodegenerative disease characterized by the accumulation of extracellular amyloid beta (Aβ) leading to the formation of senile plaques and intracellular tau aggregates that form neurofibrillary tangles (NFTs) in the brain [1][2][1,2]. Approximately, 70% of AD risk is considered to be inherited and numerous genes are frequently involved, although the actual cause and molecular mechanisms are poorly understood [2][3][4][2,3,4]. Amyloid precursor protein (APP), a transmembrane glycoprotein (type I), is the key molecular driver of AD pathogenesis. An extracellular domain and a small cytosolic domain present in APP are generally accepted to be responsible for AD progression [5]. APP is ubiquitously present in the brain and is involved in building synaptic network as well as regulating neurogenesis [6]. In addition, APP has a modulatory effect on cell surface receptors and axonal transport. However, the exact functionality of APP still remains elusive [7]. In general, upon its synthesis in the endoplasmic reticulum, APP undergoes phosphorylation and glycosylation and is finally transported into the Golgi apparatus. Additional processing of APP occurs in the trans-Golgi-network (TGN), and the highest concentrations of APP are found in the TGN under normal physiological conditions. Cleavage of APP by α-secretase produces a soluble molecule, sAPPα, within the Aβ domain [8], and APP is taken up as a cargo by the endosomal/lysosomal degradation pathway. Lysosomal degradation is a clearance mechanism required to maintain a healthy state and to prevent the accumulation of undesirable cellular waste materials. The generated peptides play an important role in synaptic plasticity and neuronal survival in the healthy state [9]. It has been reported that early-onset AD (EOAD) is usually inherited with certain autosomal dominant alleles, however, in late-onset AD (LOAD) such inheritance is unknown. While individuals bearing one inherited copy of the APOE e4 allele have a great risk of developing AD, people who inherit two copies have a greater risk of AD [10]. In some instances, EOAD is triggered through genetic mutations that are passed on from parent to child, which is usually known as early onset familial AD (FAD). Generally, FAD occurs due to mutations in presenilin 1 (PSEN1), presenilin 2 (PSEN2), and APP genes through β-secretase (BACE-1) and γ-secretase instead of α-secretase leads to unwanted assembly and accumulation of Aβ peptides in the brain,[11] [11], thereby causing AD pathogenesis [12]. Diffusible oligomers and insoluble senile plaques are formed due to the abnormal presentation of Aβ peptides, thereby resulting in higher neurotoxicity. Moreover, fibrillary plaques are found in the intracellular spaces due to the aggregation of abnormal Aβ oligomers [13]. Hyperphosphorylation of tau, aggregation of hyperphosphorylated tau to bind and stabilize microtubules, is related to its aggregation as well as the formation of neurofibrillary tangles (NFTs) and is considered as a pathological condition of AD [14]. NFTs are formed by intracellular aggregation of hyperphosphorylated tau protein in specific brain regions [15]. Collectively, the neurotoxic effects of NFTs and Aβ associated with the excessive accumulation of extracellular plaques in the brain are a hallmark of AD pathogenesis [16].
Autophagy plays a widespread role in both physiological and multiple pathological conditions, including cancer and neuronal disorders [17], and is extensively involved in the pathogenesis of AD [18]. Autophagy, a self-digesting mechanism, is an intracellular cleansing process characterized by the engulfment of malformed proteins and damaged cellular organelles by membrane-bound vesicles known as autophagosomes [17]. These autophagosomes subsequently fuse with the lysosomes to form autolysosomes resulting degradation of dysfunctional materials by lysosomal acid hydrolases [19]. Autophagy is a complex and tightly regulated enzymatic process that is largely classified into two categories: mammalian target of rapamycin (mTOR)-independent and -dependent autophagy. A defect in the autophagy-lysosomal pathway has been linked to AD, which induces the formation of toxic Aβ aggregates and causes cellular apoptosis as well as tissue and organ damage, culminating in clinical symptoms [20]. In the initial stages of AD, Aβ may induce autophagy to accelerate their removal process by employing both mTOR-independent and -dependent pathways. Progression of AD deregulates the autophagy pathway, resulting in the continuous generation of Aβ, which exaggerates both autophagy malfunction and AD [21]. In addition, both oligonucleotides and proteins, such as miRNAs, transcription factor EB (TFEB), PSEN1, Nrf2, and Beclin-1 are simultaneously impaired in the regulation of autophagy, which are meticulously interrelated in the pathogenesis of AD [22]. Therefore, it is evident that the regulation of autophagy is crucial for APP clearance and the inhibition of AD pathogenesis. Abnormal autophagy is associated with AD pathogenesis; therefore, targeting autophagy may have a profound role in AD management [23].
2. Therapeutic Action of APP Triggered by Autophagy
The incidence of AD has posed a global health burden on elderly people and is predicted to increase significantly worldwide. Considerable effort has been devoted to developing drugs for the treatment of AD, focusing on drug structures and potential molecular mechanisms of AD. An example of a generally accepted hypothesis is that reduced levels of acetylcholine causes AD in neurons. However, drugs targeting acetylcholine, the so called “cholinesterase inhibitors” could poorly improve AD [24][25][80,81]. Therefore, examining other drugs, including potential autophagy regulators might have a greater potential for the treatment of AD.
2.1. Use of Small Molecules to Modulate Autophagy in AD
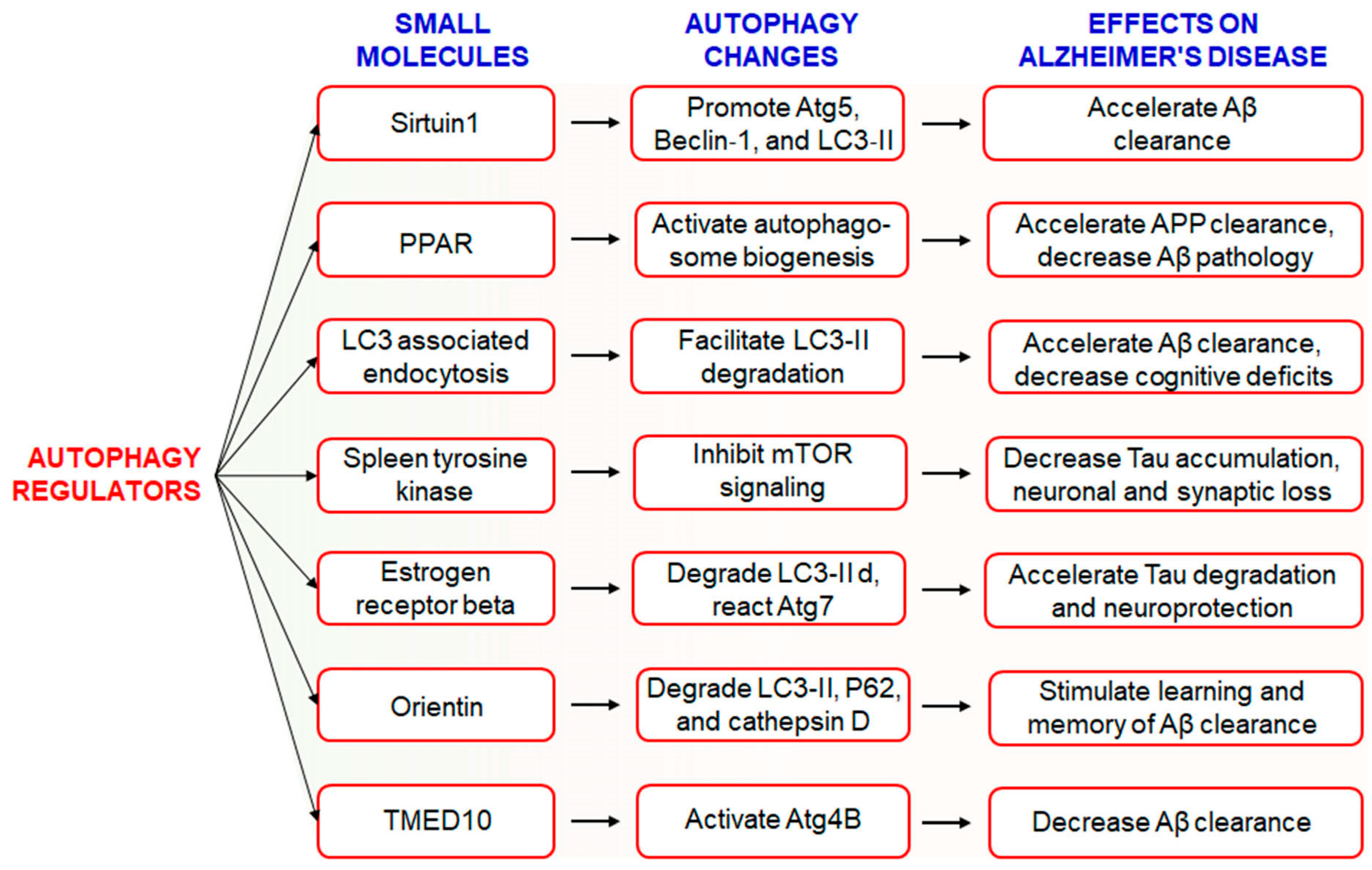
The hallmark of AD pathogenesis is the accumulation of amyloid beta proteins and hyperphosphorylated tau, which are considered toxic to neurons [26][82]. Dysregulated or insufficient autophagy might be causative factors behind the development and progression of AD [21], and therefore, the discovery of drugs targeting autophagy-related signaling pathways might be an attractive approach for the treatment and management of AD [17]. However, the Autophagy Small Molecule Database (AutophagySMDB) comprising small molecules is growing gradually. Further, an extensive database containing numerous target proteins of small molecule modulators that have curated indirect or direct evidence will be completed in the near future [27][83]. Moreover, the significance of autophagy in numerous disease states, in addition to requiring deeper examination of its molecular mechanisms. Kuang et al. has been revealed that several small molecule autophagy modulators, which may serve as prospective tools for therapeutics of AD [28][84] (Figure 1). Sirtuin1 (SIRT1), a positive regulator of autophagy, increases the expression levels of Atg5, Beclin-1, and LC3-II and accelerates the clearance of Aβ [29][85]. In addition, PPARα-mediated activation of autophagy facilitated the clearance of APP and decreased Aβ pathology in APP/PS1 mice [30][86]. Treatment with PPARα agonists decreased Aβ levels in the hippocampus and cortex, and improved autophagosome biogenesis. Together, these observations suggest that PPARα is a critical player involved in autophagy during Aβ processing [31][87]. Recently, it has been found that LC3-associated endocytosis assists Aβ clearance in addition to alleviates murine AD [88][32]. Spleen tyrosine kinase (SYK) inhibits mTOR signaling with attenuation of tau accumulation and reduces loss of synaptic function in vitro and in vivo [33][89]. Small molecule estrogen receptor β (ER) has been found to promote neuroprotective and tau degradative activity via LC3-II and Atg7 enhancement of extracellular Aβ1-42 degradation via the autophagy-lysosome system in AD [34][90]. Orientin increases learning and memory function in addition to increase clearance of Aβ via the autophagic pathway of LC3-II upregulation as well as p62 and cathepsin D degradation in an AD mouse model [35][91]. Additionally, transmembrane p24 trafficking protein 10 (TMED10) has been described to activate autophagy via ATG4B activation through decreaseing Aβ production in AD patients [36][92].
2.2. Use of Natural Compounds to Modulate Autophagy in AD
Natural products have been used to treat several neurodegenerative disorders and cancer, and have been targeted as autophagy inducers [37][93]. Recent studies have revealed that active compounds in natural products exhibit curative effects against AD via various mechanisms, including anti-cholinesterase activity, anti-apoptosis, and neuroprotective effects via anti-oxidation through targeting autophagy [38][94]. Emerging evidence suggests that natural compounds are attractive sources of autophagy regulators [39][95]. Several reports demonstrate that the active compounds regulating autophagy pave a new therapeutic approach for neurodegenerative diseases [17]. Examples of plant-derived active components that ameliorate the symptoms of AD by targeting autophagy are summarized in Table 1.
Table 1.
Modulation of autophagy by natural products and their therapeutic implication in Alzheimer’s disease.
| Natural Products | AD Model | Activities/Effects | Molecular Mechanism | References |
|---|
| Dendrobium nobile Lindl alkaloid, DNLA | Hippocampus neurons of Aβ25-35 | Protective effects of axonal degeneration | Autophagic flux enhancement | [40] | [97] | |
| Extra-Virgin Olive Oil (EVOO) | TgSwDI mice | Neuroinflammation reduction | AMPK-ULK1 pathway induction | [41] | [98] | |
| Ginsenoside Rg2 | 5×FAD transgenic mice |
Removal of Aβ aggregation |
AMPK/ULK1-mediated autophagy induction | [42] | [104] | |
| Protopanaxadiol derivative DDPU | APP/PS1 mice model | Stimulates the clearance of Aβ | Inhibition of PI3K/mTOR-mediated autophagy induction | [43] | [105] | |
| Berberine | 3×Tg-AD mice | Promotes the clearance of Aβ | Activates Bcl2/Beclin1-mediated autophagy induction | [44][45] | [101,106] | |
| Flavonoids Silibinin | Aβ1-42-induced rat model | Attenuates neuronal damage | Inhibits autophagy | [46] | [107] | |
| Corynoxine B | Tg2567 mice, N2a-SwedAPP cell model | Augments APP and Aβ degradation | Pathway that induces autophagy is unknown | [47] | [103] | |
| Gypenoside XVII | APP/PS1 transgenic mice | Prevents Aβ accumulation | Promotes TFEB to induce autophagy | [48] | [108] | |
| Ginkgo biloba | extract | TgCRND8 mice | Improves cognitive function | Induces autophagy | [49] | [109] |
| Radix polygalae | extract | Cell model of CHO-APP/BACE1 | Decreases Aβ1-40 levels | Activates AMPK/mTOR and promotes autophagy | [50] | [110] |
| Madecassoside | D-galactose-induced mouse model | Autophagy inhibition | Increases Bcl-2 and decreases Beclin-1 | [51] | [111] | |
| Hesperetin | N2a cell model | Increases Aβdamage | Autophagy inhibition | [52] | [112] | |
| Morus alba | extract | SH-SY5Y cells | Autophagy induction | mTOR-dependent autophagy pathway | [38][53] | [94,113] |
| Wogonin | SH-SY5Y-APP primary cortical astrocytes | Enhances Aβ removal | Activates ULK1/mTOR and induces autophagy | [54] | [114] | |
| Curcumin | APP/PS1 transgenic mice | Prevents Aβ deposition | Inhibits PI3K/mTOR and induces autophagy | [55] | [115] | |
| Resveratrol | N2a-APP cells, HEK293-APP cells | Decreases Aβ production and aggregation | Induces autophagy by activating AMPK/mTOR signaling | [56][57] | [116,117] | |
| Sulforaphane | AD model | Nrf2 signaling | Induces autophagy | [58] | [118] | |
| Carnosic acid | Aβ25-35-induced SHSY5Y cells | Inhibition of Aβ1-42 aggregation | Activates AMPK/mTOR and induces autophagy | [59] | [119] | |
| Tripchlorolide | 5×FAD transgenicmice | Reduces cerebral Aβ deposits | Activates PI3K/mTOR pathway | [60] | [120] | |
| β-asarone | APP/PS1 transgenic mice | Decreases Aβ level | Activates PI3K/mTOR and inhibits autophagy | [61] | [121] | |
| Oxyresveratrol | SH-SY5Y cell model | Stimulates autophagy | Atg5/7, Beclin-1, and LC-3 induction | [62] | [31] | |
| 18α-Glycyrrhetinic acid | SH-SY5Y cell model | Induction of autophagy flux | mTOR-dependent autophagy induction | [63] | [30] | |
| Gintonin | Mouse cortical Astrocytes, APPswe/PSEN-1 | Autophagic flux induction, cognition improvements | Beclin-1, Atg5/7, LAMP-1 induction, elevation of hippocampal neurogenesis | [64][65][66] | [29,122,123] | |
| Emodin | APP/PS1 mice | Autophagy inhibition | Activates Bcl-2/Beclin-1/PIK3C3 pathway | [47] | [103] |
2.3. Use of FDA-Approved Drugs to Modulate Autophagy in AD
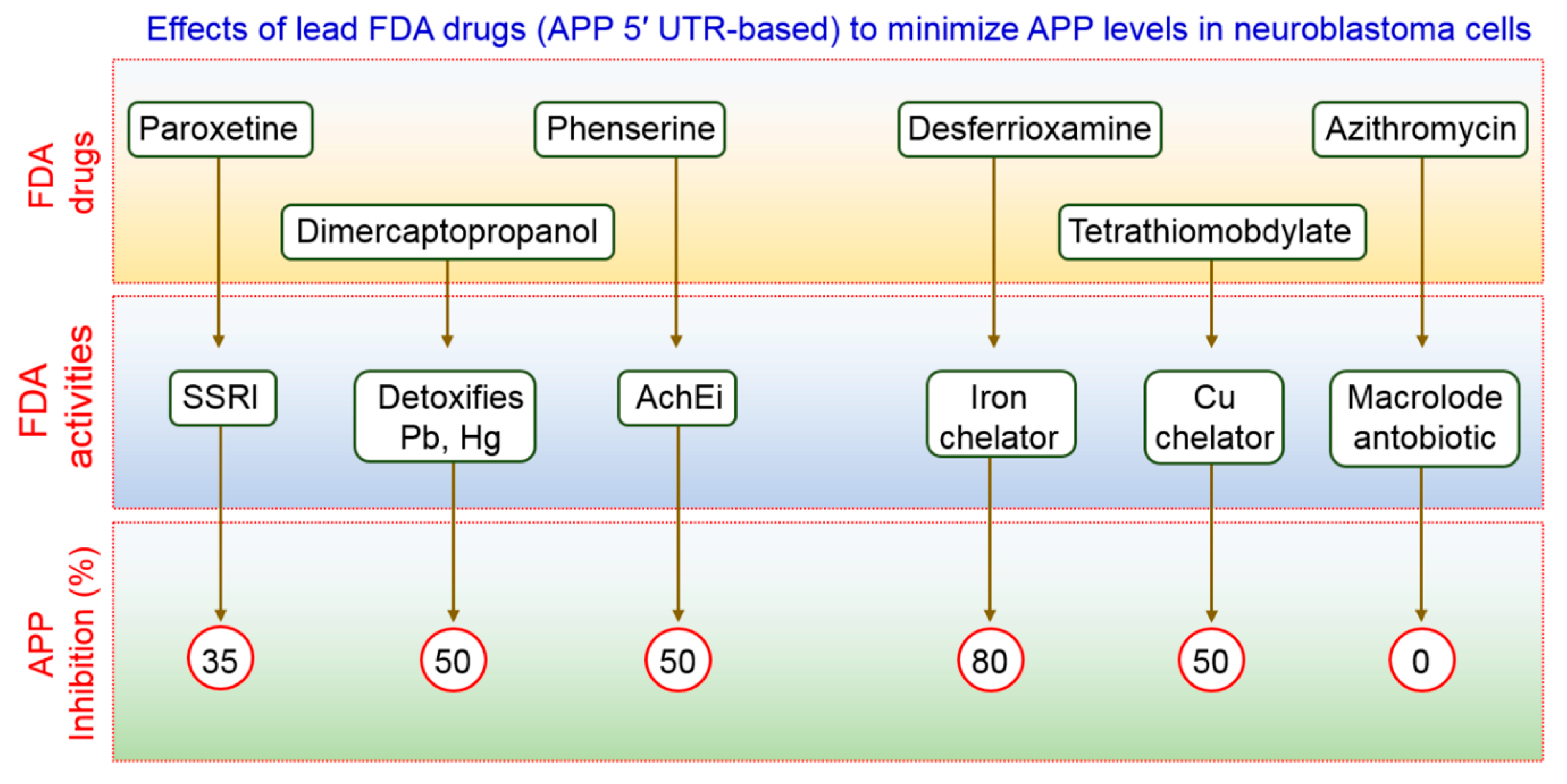
FDA-preapproved as well as approved drugs have the main advantage of being characterized fully for their pharmacological activity. These kinds of drugs have been shown to be safe in reducing toxicity, possibility of oral administration, ability to cross the BBB, and half-life are contained within pharmacologically standard. Clomipramine, an FDA-approved drug used for the treatment of psychiatric disorders, was shown to block the fusion of autophagosomes with the lysosomes, thus interfering with autophagic flux [67][131]. Another FDA-approved drug, a benzoporphyrin derivative, known as verteporfin, is likely to inhibit the formation of autophagosomes in the presence of chloroquine (CQ), a lysosomal inhibitor [68][132]. APP 5′ UTR-directed drugs decreased APP levels in SH-SY5Y neuroblastoma cells [69][133]. As shown in Figure 2, lower levels of Aβ peptides were achieved by using FDA-preapproved drugs that lowered intracellular APP holoprotein levels in SH-SY5Y cells have been demonstrated by Morse et al. [70][134]. The pharmacological action of DMP, DFO, paroxetine, phenserine, and tetrathiolmobdylate in decreasing the levels of APP and Aβ peptide is shown in Figure 2. Azithromycin dramatically changed the processing of APP [71][135]. Thus, it has been reported that a subsection of drugs that have been selected to stimulate APP 5′ UTR-mediated translation is, in addition, coactivators of the non-amyloidogenic pathway of APP processing. Therefore, it is urgently required to test an increased number as well as more sophisticated FDA-approved drugs with relative effectiveness in larger group of animals to modulate autophagy in AD pathogenesis.