Obesity has become a global epidemic and a public health crisis in the Western World, experiencing a threefold increase in prevalence since 1975. High-caloric diets and sedentary lifestyles have been identified as significant contributors to this widespread issue, although the role of genetic, social, and environmental factors in obesity’s pathogenesis remain incompletely understood. In recent years, much attention has been drawn to the contribution of the gut microbiota in the development of obesity, driven by a body of evidence supporting its central role in regulation of host physiology. Indeed, research has shown that in contrast to their healthier counterparts the microbiomes of obese individuals are structurally and functionally distinct, strongly suggesting gut dysbiosis as a key factor underpinning an obese phenotype. The following articlentry explores the myriad of mechanisms by which the microbiome may contribute to the etiology of obesity, including modulation of host energy balance, immune function and macronutrient metabolism.
- microbiome
- obesity
- metabolic syndrome
1. Introduction
The complex ecosystem of the gut microbiota comprises over 1000 unique bacterial strains, with a prokaryotic population that outnumbers the total cells in the human body by an order of magnitude[1] [8]. Only recently has research begun to unravel the intricate links between the microbiome and human health, especially in the context of host metabolism. Just like other organs in the body, the microbiome can dynamically respond to a variety of internal and external physiological cues, such as food intake, energy requirements, and stress, in order to maintain a state of metabolic homeostasis. Accordingly, perturbances to the microbiome that lead to an unstable or “dysbiotic” state are linked to the pathophysiology of various metabolic conditions, including obesity.
The first studies establishing a causal link between the microbiome and an obese phenotype were performed in germ free (GF) mice, which were initially found to be resistant to diet-induced obesity even under conditions of high-fat (HF) feeding[2] [15]. Furthermore, an obese phenotype was transmissible to these animals via fecal transplant from either Western diet-fed or genetically obese mice, which induced greater weight gain than inoculation with wild-type microbes[3] [9]. Similar results have been observed in colonization studies with microbiota from pairs of mono and dizygotic human twins discordant for obesity. Specifically, microbial inoculation leads to a progressively greater increase in fat mass and body weight in animals receiving microbes from the obese twin, despite no significant differences between animal groups in energy intake[4] [16].
Inspired by these findings, a multitude of microbial survey studies have attempted to define what constitutes an “obese” microbiome, and pinpoint the specific strains that contribute to development of obesity and related metabolic symptoms. Based on such investigations, a lean phenotype has largely come to be associated with an increased Bacteriodetes:Firmicutes ratio, whereas this taxonomic proportion is inverted in obese individuals[5][6] [17,18]. Further supporting this model, studies monitoring the microbiome of obese patients during weight loss have observed a “remodeling” of the gut ecosystem to contain a higher relative amount of Bacteroidetes subtypes[7][8] [19,20]. However, further human studies and meta analyses have revealed this one-to-one association may not be as unambiguous as previously thought. Notably, a recent metanalysis by Sze et al. found a negligible association between the Bacteriodetes:Firmicutes ratio and obesity status across analytical studies, and that high levels of experimental “noise” in the form of interpersonal variation and small sample size likely confounded these early observations[9] [21]. This, along with other inconsistency in the literature, suggests that salient differences in pathology-related microbiota may occur at a more precise phylogenetic level than the broad divisions described above.
Although it remains unclear the exact taxonomic composition that constitutes a “healthy” gut microbiota, it is evident that microbial diversity is an essential component to host health. Compared to their lean counterparts obese individuals have a markedly lower bacterial diversity, and decreased fecal microbial gene richness is associated with various physiological markers of obesity and metabolic syndrome[10] [22].
Building on this notion, other studies have argued for the importance of a healthy “core” microbiome, comprising the diverse bacterial genes required for metabolic integrity, that is perturbed in the clinically obese[11] [23]. This diversity-centered model aligns well with the plethora of metabolic niches occupied by the microbiome, supporting various homeostatic processes that are critical to host health. It thus follows that in absence of sufficient microbial diversity to sustain these functions, or presence of a dysbiotic gut microbiota with perturbed metabolic capacity, development of a pathological state may occur. Indeed, a normal healthy microbiota may be better defined at a functional rather than at a compositional level, such that certain essential bacterial species can dynamically respond to changes in host metabolism.
In the case of obesity and metabolic syndrome, it is important to understand not only the microbiome’s contribution to metabolic function in healthy individuals, but how perturbations to the microbiome may promote a diseased state (see Figure 1). However, the interplay of these mechanisms and how they influence the overall metabolic status of an individual has yet to be fully understood. Thus, a discussion of how the microbiome can be targeted in treatment of obesity first warrants examination of the unique channels mediating host–microbiome interactions, and the evidence for their involvement in metabolic pathology.
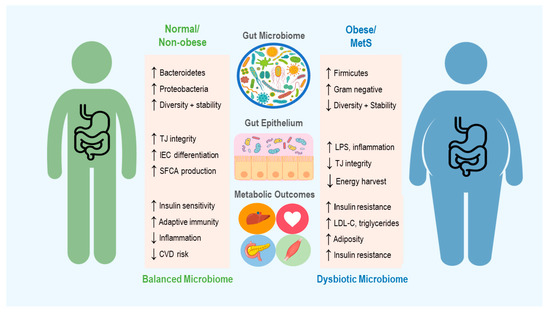
Figure 1. An overview of the microbiome’s role in development of obesity and metabolic syndrome (MetS), including some of the mechanisms thought to contribute to changes in host metabolic state. Up and down arrows indicate increase and decrease, respectively. TJ = Tight Junction, IEC = Intestinal Epithelial cells, SCFA = Short Chain Fatty Acid, CVD = Cardiovascular Disease, LPS = Lipopolysaccharide and LDL-C = Low Density Lipoproteins-Cholesterol.
2. The Microbiome in Energy Harvest and Expenditure
Despite the complex genetic, social, and environmental parameters contributing to its development, the core etiology of obesity depends on a chronic positive energy balance. More precisely, dysregulation of nutrient partitioning in a state of perpetual energy surplus leads to fat storage and weight gain, with a myriad of associated disturbances to organ and tissue function[12] [24]. The intestinal microbiota modulates energy balance by extracting calories from indigestible carbohydrates in the human diet, which are fermented into short chain fatty acids (SCFAs) and other metabolites. These by-products of microbe metabolism can subsequently serve as a form of bioavailable fuel for cellular processes in various tissues and organs. Indeed, colonocytes obtain 60–70% of their cellular energy from SCFA oxidation[13] [25], and the fraction of substrate not consumed by the colon epithelium is transported into systemic circulation such that it can be mobilized to peripheral tissues[13] [25]. It is estimated that through this energy extraction paradigm SCFAs provide ≈10% of daily caloric requirements in humans[14] [26]. Different species of SCFA can also have different metabolic fates: whereas propionate is primarily a precursor for gluconeogenesis, acetate and butyrate are preferentially incorporated into fatty acids and cholesterol[15] [27]. Furthermore, in addition to an important energy source SCFAs serve as active signaling molecules, that interact with G-protein coupled free fatty acid receptors (FFARs) in various tissues to exert a broad spectrum of effects on lipid, glucose, and protein metabolism.
An individual’s intestinal SCFA profile relies on a variety of endogenous and external factors, including abundance of fermentable substrates, host–microbiome interactions, host lifestyle, and gut bacterial diversity. In turn, composition and size of the SCFA pool is an important determinant of host metabolic state[16] [10]. The involvement of SCFAs and energy harvest in obesity was first brought to light in seminal studies performed by Bäckhed et al., in which GF mice were protected against diet induced obesity compared to their wild-type littermates[2] [15]. In keeping with this result, GF mice also displayed reduced concentrations of intestinal SCFAs[17] [28] and doubled their caloric excretion of undigested polysaccharides in feces and urine compared to conventional animals[18] [29], supporting the causal role of gut bacteria in converting these substrates into a bioavailable energy source for the host. Furthermore, colonization of these mice with a healthy microbiome was sufficient to increase intestinal SCFAs and induce adiposity[2] [15], while colonization with microbiota from an obese donor doubled this subsequent gain in fat mass[3] [9]. Many animal and human studies have since confirmed increased levels of cecal and fecal SCFAs in obese subjects compared to their lean counterparts, indicative of higher rates of carbohydrate fermentation and energy extraction[19][20][21][22] [30–33].
Further studies by Turnburgh et al. revealed that the microbiome of obese (ob/ob) mice displayed an enrichment of bacterial genes associated with increased energy harvest[3] [9]. This finding has been confirmed in humans, via comparison of the microbial transcriptome in obese and lean di/monozygotic twins. Similar to obese mice, the obese human gut microbiome is enriched for genes involved in microbial processing of carbohydrates, an association representative of taxonomic differences in Actinobacteria (contributing 75% of obese-enriched genes) and Bacteroidetes (contributing 42% of lean-enriched genes)[11] [23].
However, it appears that the metabolic effects of SCFAs are complex and pleiotropic, simultaneously conferring many benefits to the host that counteract their contribution to energy surplus and adipogenesis. For example, while G-protein coupled receptor (GPCR) mediated SCFA signaling serves to increase colonic transit time which may further enhance bacterial fermentation and energy extraction, it can also contribute to absorption of vital micronutrients from ingested food and promote host bowel regularity[23] [34].
This dual role of SCFAs may in fact provide a net benefit, as multiple studies have demonstrated a function for SCFAs in protecting against diet-induced obesity. The reduction of weight gain by SCFA metabolites has been linked to a variety of mechanisms, including modulation of metabolic flux and satiety signaling. For one, SCFA signaling through FFARs 2 and 3 stimulates the secretion of glucagon-like peptide 1 (GLP-1) from intestinal cells and promotes intestinal gluconeogenesis, pathways that act twofold to enhance insulin sensitivity and reduce appetite[24][25] [35,36]. Furthermore, in both mouse and human studies SCFAs have been shown to enhance intestinal production of the anorexigenic peptide YY (PYY) and the adipocyte-associated hormone leptin, both of which increase satiety levels and promote reduction of energy intake[23][26][27][28][29] [34,37–40].
In addition to these enteroendocrine pathways, recent work has also demonstrated the neuroactive properties of SCFA metabolites, allowing direct modulation of appetite control. Particularly, acetate can cross the blood–brain barrier and enhance hypothalamic GABAergic neurotransmission, repressing appetite and reducing energy intake[30] [41] Similarly, butyrate can suppress activity of orexigenic neurons in the hypothalamus and vagal afferents in the brainstem, an effect shown to mediate reduced food intake and protection against the effects of high-fat feeding[31][32] [42,43].
Complementary to reducing food intake, many animal studies have confirmed the positive influence of SCFAs on body weight through increasing host energy expenditure. This effect is associated with upregulation of various thermogenic genes, thereby leading to enhanced mitochondrial function, browning of adipose tissue, and activation of lipid oxidation via gut–neural signaling pathways[33][34][35][36] [44–47]. Specifically, den Besten et al. showed that SCFA supplementation upregulates expression mitochondrial uncoupling protein 2 and raises AMP-to-ATP ratio, thereby stimulating oxidative metabolism in liver and adipose tissue via AMPK[36] [47]. The phenotypic outcome is dramatic increases to energy expenditure as well as reduced body weight and fat mass, despite little change in nutrient intake or high-fat feeding.
These effects have been investigated in humans, with similar overall outcomes despite providing less mechanistic insight. Canfora et al. showed that colonic administration of SCFA mixtures (comprising acetate, propionate, and butyrate) increased fasting lipid oxidation and resting energy expenditure (REE) in overweight and obese subjects[37] [48]. Further in vivo data from human cohorts has corroborated the finding that SCFAs raise whole-body REE and lipid oxidation[38] [49], and shown these changes to be independent of fasting glucose and insulin levels[39] [50]. Although mechanistic studies of these SCFA-induced effects are lacking in humans, current work suggests they provide significant improvements to oxidative metabolism that may translate into long-term benefits in weight control.
3. Regulation of Immune Function
One of the hallmarks of obesity and metabolic syndrome is a systemic, low-grade inflammatory state. Research has shown that a wide range of inflammatory markers, including C-reactive protein and pro-inflammatory cytokines, are strongly associated with development of adiposity[40][41] [51,52] and increased risk of metabolic disorders such as cardiovascular diseases, fatty liver disease, and type 2 diabetes[42] [53]. More recently, direct mechanistic links have been suggested between obesity-associated systemic inflammation and the development of insulin resistance, the core diagnostic symptom of metabolic syndrome[43][44] [54,55].
Although the causal relationships linking obesity, metabolic syndrome, and inflammation are incompletely understood, multiple lines of evidence implicate dysbiotic gut microbiota as a key modulator of immune signaling in the context of metabolic pathology. For one, lipopolysaccharide (LPS) derived from the cell wall of pathogenic Gram-negative microbes can bind toll-like receptors (TLRs) in mucosal and peripheral tissues, initiating pro-inflammatory signaling cascades[45][46] [56,57]. Data from both human and rodent studies has linked an obese phenotype to elevated circulating levels of plasma LPS, a condition known as metabolic endotoxemia[47][48][49] [58–60]. For example, in a comparative study of human subjects, baseline circulating endotoxin levels were found to be 20% higher in obesity or glucose intolerant individuals and 125% higher in type 2 diabetics compared to lean subjects[50] [61].
Rodent studies by Cani et al. were the first to causally link metabolic endotoxemia to an obese phenotype. They found elevated plasma LPS could be induced by high fat feeding, which subsequently led to increased adiposity and metabolic dysregulation in the form of hyperglycemia and insulin resistance[51] [62]. Interestingly, however, the same effect was achieved via artificial subcutaneous infusion of LPS into the blood plasma, even in absence of high fat feeding. Thus, not only is diet a direct factor in modulating systemic inflammation, but a pro-inflammatory state is sufficient to promote obesity and perturbed metabolic function. More recent studies have mechanistically corroborated these findings, linking microbiota-related inflammatory changes during HFD-induced obesity to Toll-like receptor 4 (TLR4) signaling and a resultant increase in plasma levels of LPS[52] [63].
The potential contribution of the gut microbiome, especially a perturbed microbiome as seen in the context of obesity, to this phenomenon is twofold. For one, pathogenic strains that may dominate a dysbiotic gut are a rich source of LPS and other endotoxins, that may infiltrate circulation to initiate an immune response[45][46] [56,57]. Second, there is strong evidence for the critical role of the gut microbiota in maintaining integrity of the gut epithelial lining, a function that if compromised would permit increased intestinal translocation of endotoxins into the blood[53] [64].
The gut microbiome occupies the outer mucus layer of the intestinal epithelium, where it can interact with the luminal environment and metabolize dietary components. The inner mucus layer of the gut epithelium, on the other hand, is critical for limiting the exposure of epithelial cells to the microbiome and other potential pathogens entering the lumen from the external environment. However, resident bacteria also serve as a crucial line of resistance to colonisation and invasion by exogenous microbes that may harm the host[54] [65]. Thus, the symbiotic yet complex relationship between the microbiome and gut epithelium serves to maintain a robust and tightly regulated mucosal immune defense mechanism.
Unlike pathogenic strains, many commensal species of gut microbes are known to help stabilize the mucosal membrane through promoting regular turnover of mucin glycoproteins[45][46] [56,57] and stimulating intestinal endocannabinoid production that may help attenuate inflammation[55] [66]. Furthermore, exposure to probiotic microbial species has been shown to promote upregulation of intracellular tight junction proteins[56][57] [67,68] that provide an essential structural framework for maintaining mucosal barrier function.
Additional work has also pinpointed the role of bacterial SCFAs in maintenance of gut epithelial immunity. In vivo studies have demonstrated the potent trophic effects of SCFA metabolites on colonic epithelium cells. These include providing energy for cell growth, stimulating epithelial cell proliferation and differentiation, and enhancing mucus secretion, all functions that normalize intestinal permeability[58][59] [69,70]. This not only further emphasizes the importance of bacterial SCFAs in promoting gut health, but provides mechanistic explanation for the observation that SCFA administration decreases systemic inflammation and immunoreactivity [60][71].
Thus, when considering the role of the gut microbiota in the etiology of obesity, one can attribute significant weight to the impaired intestinal barrier integrity and subsequent metabolic endotoxemia that develops in the presence of a dysbiotic microbiome structure. It follows that restoring a normal, healthy equilibrium between resident microbes and innate mucosal immunity would attenuate these systemic effects and potentially preclude development of morbid adiposity.
4. Regulation of Bile Acid Metabolism
The gut microbiota also participates in various stages of bile acid (BA) metabolism, and the bidirectional crosstalk between hepatic BA production and microbial ecology mediates a key interaction between the microbiome and the host. Postprandial primary BAs (cholic acid and chenodeoxycholic acid) released by the liver are modified and metabolized by the gut microbiota to produce bioactive secondary BAs, including deoxycholic acid (DCA) and Lithocholic acid (LCA)[61] [72]. These microbial metabolites can bind cellular receptors including farnesoid X receptor (FXR) and Takeda G protein-coupled receptor (TGR5), thereby exerting profound downstream effects on lipid metabolism, cholesterol balance, and insulin sensitivity[62] [73] that are particularly relevant to the discussion of metabolic pathology.
The dysbiotic microbiome structure displayed in obese phenotypes is accompanied by altered BA pool composition and metabolism, and a number of studies have associated elevated BAs and secondary BAs with chronic conditions including obesity and type 2 diabetes[61] [72]. It has been shown that high fat feeding results in adiposity as well as an increase in total BAs across tissues (particularly deoxy- and taurodeoxycholic acid), which is associated with taxonomic microbiome restructuring to favor strains capable of BA processing[63] [74]. Interestingly, another important property of BAs is that they exert potent anti-microbial effects, providing a feedback mechanism by which BA levels exert a strong selective pressure on the microbiota[64] [75]. In fact, direct cholic acid supplementation has been shown to produce similar changes in microbiome composition to those seen in diet-induced obesity, including an expansion of members in the class Firmicutes capable of DCA production[64] [75]. Thus, BA transformation by the gut microbiota can initiate changes in the BA pool size, while BAs can conversely initiate changes in the diversity and composition of the gut microbiota, both of which dramatically impact host physiology.
The aforementioned signaling properties of select primary and secondary BAs further complexifies this axis of microbe–host communication, especially in the context of obesity and metabolic disease. On one hand, BA-dependent TGR5 activation on the surface of enteroendocrine cells increases secretion of the incretin hormone GLP-1, which improves glycemic regulation in liver and pancreas, protects against insulin resistance, and improves satiety[65] [76]. In addition, stimulation of TGR-5 by BAs induces browning of adipose tissue and increases skeletal muscle energy expenditure through thyroid hormone signaling, protecting against diet-induced obesity[65] [76].
The role of FXR-mediated BA signaling in obesity is markedly more pleiotropic and complex, and is impacted by the microbiome via multiple avenues. For one, negative regulation of BA production by FXR signaling provides a mechanism by which the microbiome may directly influence the size of the BA pool and regulate lipid homeostasis[66] [77]. This was demonstrated in elegant studies by Sayin et al. who found that in addition to regulating secondary BA metabolism the microbiome also inhibits BA synthesis in the liver by alleviating FXR inhibition in the ileum[67] [78]. Furthermore, BA agonism of FXR can initiate the release of fibroblast growth factors (FGFs) 19 and 21, both of which contribute to insulin sensitization and hypolipedmia[68] [79]. Finally, the bacterial enzyme bile-salt hydrolase (BSH) relieves inhibition of FXR signaling via selective cleavage of its antagonist tauro-β-muricholic acid (TβMCA). Colonization of mice with microbiota displaying enhanced BSH activity leads to reduced body weight, as well as lower serum cholesterol and hepatic triglycerides in colonized GF mice[69] [80].
Although these findings may suggest a positive role for FXR in body weight homeostasis, other research suggests otherwise. Most notably, FXR-deficient mice are actually protected against both diet-induced obesity as well as induction of an obese phenotype via fecal transplant[70] [81]. Furthermore, mice colonized with an obese microbiome show increased ileal BAs and FXR mRNA, suggesting a role for increased FXR signaling in microbial transmission of an obese phenotype[4] [16]. Finally, and perhaps most interestingly, in direct contradiction to the aforementioned study Yao et al. found that knockout of B. thetaiotaomicron BSH in gnotobiotic mice led to reduced weight gain on a high-fat diet compared to WT-colonized mice[71] [82]. This effect was associated with higher in-vivo levels of TβMCA, suggesting that repression of FXR signaling is key in preventing diet-induced adiposity.
Overall, although the exact signaling mechanisms connecting microbiome activity, secondary Bas, and obesity have yet to be entirely described, it is clear that phylum-specific regulation of host BA metabolism has a direct and profound impact on the development of pathological adiposity and related complications in glucose and lipid metabolism.
5. Role of the Diet in Shaping the Microbiome
As research continues to corroborate the central importance of the microbiome in host health and pathological processes, modulating the microbiome to potentially rectify dysbiotic metabolic states has become a research area of great interest. Among the factors contributing to microbiome dynamics, it has become well established through mouse and human studies that diet can play a critical role in microbiome remodeling and dramatically alter its structure within a mere 24 h. Administration of a Western diet promotes restructuring of the distal gut microbial community such that a Mollicute lineage in the Firmicutes, normally present at low abundance in the mouse colon, expands dramatically to dominate this body habitat[72] [83]. This effect may be largely due to the competitive metabolic advantage conferred upon these strains by an abundance of simple sugars, such as sucrose, allowing their expansion to dominate other microbial sub-populations[72] [83]. In addition, a high-fat, obesogenic Western diet has been proven to significantly reduce bacterial diversity and richness in the GI tract of mice, an effect that is readily reversible upon reverting back to a normal chow diet[73] [84]. This effect has also been replicated in humans transitioning between high-fiber and high-fat-and-simple-sugar diets, with the microbiome showing equally flexible functional and taxonomic profiles[74] [14].
Interestingly, the deleterious effect of HFD was also demonstrated to be compounding in nature, as persistent microbial signatures during repeated cycles of HFD lead to enhanced metabolic derangements and accelerated weight gain after a period of normal feeding[75] [85]. Expanding on this finding, work by Sonnenburg et al. has shown that such diet-driven changes in microbiota may span further than an individual’s lifetime, inducing extinction of certain commensal strains across generations[76] [86]. Restoration of diversity and reappearance of specific microbes in this context could only be achieved upon fecal microbial transplant (FMT) but not from diet switching.
Overall, these data suggest that the metabolic perturbances correlated with the poor Western nutrition may in fact be due to diet-induced insult to our gut microbiome. In light of this, modulating the microbiome via dietary intervention offers a unique therapeutic strategy that may not only serve to reintroduce beneficial strains, but also sustain restoration of a healthy microbial ecosystem.