Parkinson's disease (PD) is the second most common neurodegenerative disease, affecting 1% of the population over 60 years of age. Clinically, PD is characterized mainly by the presence of motor symptoms, but a wide range of non-motor symptoms not only accompany the disease, but many of them even prevent the occurrence of motor symptoms. Despite many efforts, current medicine fails, not only in the treatment of PD, but in the early diagnosis of the disease. Current treatment is mainly based on symptomatic treatment or treatment with levodopa (L-DOPA) but does not focus on the pathogenesis of the disease itself. The presence of non-motor symptoms such as gastrointestinal dysfunction, which occurs up to 20 years before the onset of motor symptoms themselves, provides a sufficiently wide window of early diagnosis and thus potential early therapy for these patients.
α-synuclein (α-Syn) is one of the candidates for a biomarker of PD. Mutations in its gene are associated with the familial form of PD as well as conformational changes in the protein and with its ability to oligomerize and fibrillate and form Lewy bodies (LB) and Lewy neurites (LN). It is LB and LN that are considered to be one of the most important pathological features of the disease, where α-Syn is the major component of these pathological structures. Its direct association with the pathogenesis of the disease together with the fact that in addition to brain structures, α-Syn is present in body fluids as well as in peripheral tissues such as gastrointestinal (GIT) tissues even enhances its potential use in early diagnosis of the disease.
The diagnosis of the disease is also complicated by the similarity of clinical, cognitive, and neuropathological features of PD with other neurodegenerative diseases. In this context, various α-Syn strains have been identified based on the ability of α-Syn to form several types of fibrillar structures (strains) with different conformational, biological, and biochemical properties. And it is these differences that may be key to the correct early diagnosis of PD.
- Parkinson`s disease
- alpha-synuclein
- alpha-synuclein strains
1. Introduction
Parkinson’s disease is (PD) the second most common age-related neurodegenerative disease after Alzheimer’s disease (AD). PD affects approximately 1% of the population over the age of 60, with an increasing incidence with age [1,2][1][2]. Clinically, the disease is mainly characterized by the presence of motor symptoms, such as bradykinesia, postural instability, and resting tremor [3]. However, the presence of non-motor symptoms such as olfactory disorders, gastrointestinal (GI) dysfunction, autonomic failure, cognitive disorders, mental health problems, sleep disorders, pain, and excessive fatigue, not only accompany this disease, but many of them even precede the occurrence of motor symptoms [4,5][4][5]. It is the occurrence of non-motor symptoms, such as GI dysfunction, which occur up to 20 years before the onset of motor symptoms themselves [6[6][7],7], that provides a wide window in terms of early diagnosis, and thus potentially therapy for these patients. This is exacerbated by the fact that current therapy is based mainly on symptomatic or levodopa (L-DOPA) treatment but does not target the pathogenesis of the disease itself. Despite intensive efforts, the development of new and more successful therapeutic approaches has been insufficient, and fully dependent on early diagnosis of the disease. However, the diagnostics, especially in the early stages of the disease, currently mainly rely on very non-specific clinical symptoms, such as olfactory disorders, GI dysfunction, mood, and sleep disturbances. Whereas, the definitive confirmation of the disease is still based on a post-mortem pathological evaluation of the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), and the presence of aggregated protein in a neuronal soma called the Lewy body (LB) and in neuronal processes called Lewy neurites (LN) [8,9,10][8][9][10]. Another complication in the diagnosis is the similarity of the clinical, cognitive, and neuropathological signatures of PD with other neurodegenerative diseases, such as PD with dementia (PDD), dementia with LB (DLB), AD, multiple system atrophy (MSA), or progressive supra nuclear palsy (PSP) [8,11,12,13,14,15,16][8][11][12][13][14][15][16]. Due to these similarities, up to 20–35% of patients are misdiagnosed with PD, while not meeting strict clinical or histopathological criteria [8,17,18,19][8][17][18][19]. For example, cognitive disorders that occur in 10–80% of cases of PD, especially in the later stages of the disease, significantly overlap with PDB, DLB, AD, and MSA [8]. However, in contrast to PD, in the case of DLB and PDD, motor symptoms appear concurrently or later, after the onset of cognitive impairment [20]. In the case of histopathological signatures, typical spherical LB and LN occur in PD, DLB, and PDD [20,21][20][21], while accumulated α-Syn in MSA is localized in oligodendroglia or glial cytoplasmic inclusions, without LB with typical spherical structures [22]. It is MSA that is often misdiagnosed as PD, especially in the early stages of the disease. However, the disease is more progressive compared to PD, and the response to dopaminergic treatment is weaker [23].
2. Diagnostic Potential of α-Syn
α-Syn, as the major component of LB and LN, is consistent with the pathogenesis of PD and represents one of the possible candidates for a suitable biomarker [25][24]. In addition, mutations in SNCA gene (A53T, A30P, E46K, H50Q, G51D), its duplications, and triplications have been found in familiar PD cases. These mutations are associated with the induction of oligomerization and fibrilization of α-Syn, and thus with the development of PD [20,26,27][20][25][26]. Similarly, polymorphism in SNCA, or its promoter, poses an increased risk of disease [28,29][27][28]. On the other hand, it should be borne in mind that LBs are present in brainstem nuclei in about 8–12% of clinically asymptomatic populations over 60 years of age, which is thought to be a pre-symptomatic stage of the disease [8,30][8][29].
α-Syn is a small (140 aa), a cytoplasmic protein, highly expressed in the brain, where it represents 1% of the total protein. This high expression in brain structures, as well as its localization predominantly in the neuronal presynaptic terminals, and the ability to interact with membrane structures, predetermines its function in modulating neuronal membrane stability and regulating synaptic function, and vesicular transport [4]. In addition to brain structures, α-Syn is also present in body fluids (cerebrospinal fluid (CSF), plasma, blood) [8,26,31,32,33][8][25][30][31][32] and in peripheral tissues (skin, GI tract (GIT), olfactory glands, gonadal tissues) [14,34,35,36,37,38][14][33][34][35][36][37].
3. Structure, Posttranslational Modifications, and Toxic Forms of α-Syn
α-Syn is a cytoplasmic protein, naturally occurring as an unstructured monomer or metastable tetramer, which is able to adopt a wide range of dynamic structures, depending on the environment and binding partners [39,40,41][38][39][40]. Several different isoforms of α-Syn have been identified, including truncated variants (112, 126, and 98 aa) possessing a higher risk of pathological α-Syn aggregation [42][41].
The N-terminal and central regions of the protein (1–95 aa) are responsible for its interaction with membrane structures, whereas interaction with lipids is thought to play a role not only in modulating its function but also in changing its conformation from unstructured to highly organized fibrils [20,40,43][20][39][42]. In this regard, the central NAC region (non-amyloid-β component, 60–95 aa), as the most hydrophobic part of the protein, is able to undergo a conformational change from random coil to β-sheet structure, and form cylindrical β-sheets and amyloid-β-like fibrils [44,45,46][43][44][45]. It is the process of nuclear-dependent gradual α-Syn oligomerization and fibrilization that is considered critical in the development of PD, with the highest rates of cytotoxicity observed especially in unstable oligomeric or protofibrillar forms of α-Syn [20,26,47][20][25][46]. In addition, PD-associated point mutations are located only in the N-terminal region (A53T, A30P, E46K, H50Q, G51D) [47] [48] (Figure 1).
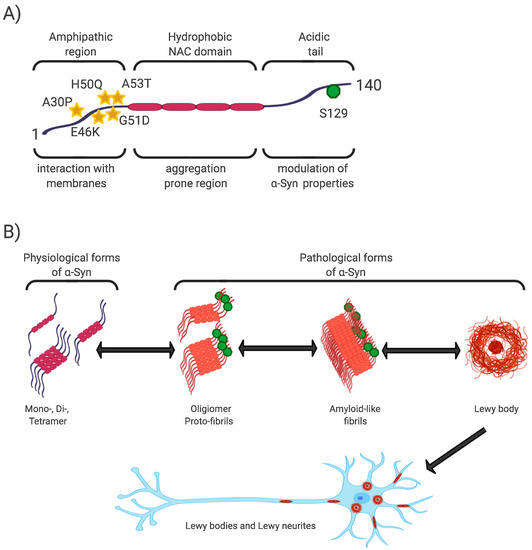
Figure 1. α-Syn structure, properties, and pathological forms. (A) Simplified scheme of α-Syn structure. α-Syn is a 140 aa protein, where the N-terminal and non-amyloid-β component (NAC) regions are responsible for its interaction with membrane structures. Mutations associated with Parkinson’s disease (PD) are located in the N-terminal region. The NAC region, the most hydrophobic part of the protein, is involved in random-coil conformational changes on the β -sheet. The unstructured C-region is responsible for the structural and physiological properties of the protein. The most discussed post-translational conditioning, phosphorylation at S129, is located in the C-terminal domain. (B) α-Syn is a cytoplasmic protein that occurs naturally as an unstructured monomer or metastable tetramer [49][48]. Under pathological conditions, its conformational changes occur from a random-coil to a beta-sheet structure to form unstable oligomers and protofibrils, followed by fibrils until insoluble protein aggregates are formed. These are localized in the neuronal soma (Lewy body) or in the neuronal processes (Lewy neurites).
The C-terminal region (96–140aa) is present in all conformational forms of the protein (monomeric, fibrillar) or in its membrane-bound form, and is responsible for the internally disordered nature of α-Syn [50,51,52,53][49][50][51][52]. It is involved in the modulation of α-Syn properties, such as interactions with other proteins [54,55[53][54][55],56], metal ions [57][56], and other ligands [45,58][44][57]. In addition, this area is rich in regulatory posttranslational modifications (PTM), which are likely to play an important role in the regulation of α-Syn structural and physiological properties [52,59][51][58] (Figure 1).
4. The Role of α-Syn Phosphorylation in the Development and Diagnosis of PD
Phosphorylation at S129 (p-S129) is the most discussed PTM in α-Syn, its role in modulating α-Syn physiological function and promoting or suppressing α-Syn toxicity is still unclear [51]. In vitro and in vivo studies have confirmed the importance of p-S129 in the regulation of (i) the interaction of α-Syn with membrane structures, with p-S129 showing an inhibitory effect in this respect; (ii) protein-protein interactions; (iii) synaptic plasticity, although the mechanism remains unexplained; (iv) α-Syn nuclear translocation; (v) interactions with metal ions, thereby affecting metal ions-mediated α-Syn aggregation properties; and last but not least, (vi) turnover, where p-α-Syn is considered to represent a late stage in the pathogenesis of α-Syn, and its main role is to selectively target proteins for their degradation [51][58][59].
5. The α-Syn Strains
As mentioned above, ß-sheet-rich α-Syn fibrils occur in several neurodegenerative diseases (PD, PDD, DLB, MSA, and AD). However, these diseases possess different clinical manifestations as well as pathological signatures [61][60]. The fact that α-Syn is able to form several types of fibrillary structures (strains), with different conformational properties in vitro and different biological activities, suggests that various types of strains are present in the brain of patients with different neurodegenerative diseases, and probably responsible for various clinical manifestations of these diseases [61,62,63,64,65][60][61][62][63][64]. This is also supported by a recent study, where in transgenic TgM83 mice, various α-Syn strains, either in vitro generated or isolated from brain-derived aggregates, targeted different brain regions and cell types in these mice but also induced different motor, pathological, and biochemical phenotypes [64][63]. In a more detailed characterization, using cell models but also transgenic animals, the authors were able to define different α-Syn strains based on a wide range of parameters, such as biochemical properties, including resistance to protease degradation denaturation by chaotropic agents; biological stability; biological activity, including inhibition of proteasome activity; cell infectivity and incubation period; pathological lesion tropism; and neurological features in transgenic animals [64,66] [63][65](Figure 2).
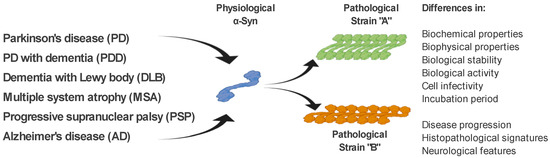
Figure 2. α-Syn strains and their discrimination. Under pathological conditions, different types of α-Syn aggregates (strains), with specific biochemical, biophysical, and biological properties, occur in the brain of patients with various neurodegenerative diseases. It is the different properties of these strains that are responsible for the different clinical manifestations of these diseases. Differential diagnosis of PD and related neurodegenerative disorders is currently quite challenging. We propose several methods to discriminate such disorders, based on the specific physical and biochemical properties of α-Syn strains.
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909.
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535.
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. Vienna 2003, 110, 517–536.
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 318–324.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418.
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87.
- Sollinger, A.B.; Goldstein, F.C.; Lah, J.J.; Levey, A.I.; Factor, S.A. Mild cognitive impairment in Parkinson’s disease: Subtypes and motor characteristics. Parkinsonism. Relat. Disord. 2010, 16, 177–180.
- Aarsland, D. Cognitive impairment in Parkinson’s disease and dementia with Lewy bodies. Parkinsonism. Relat. Disord. 2016, 22 (Suppl. 1), S144–S148.
- Aarsland, D.; Kurz, M.W. The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 2010, 20, 633–639.
- Aarsland, D.; Sharp, S.; Ballard, C. Psychiatric and behavioral symptoms in Alzheimer’s disease and other dementias: Etiology and management. Curr. Neurol. Neurosci. Rep. 2005, 5, 345–354.
- Fayyad, M.; Salim, S.; Majbour, N.; Erskine, D.; Stoops, E.; Mollenhauer, B.; El-Agnaf, O.M.A. Parkinson’s disease biomarkers based on alpha-synuclein. J. Neurochem. 2019, 150, 626–636.
- O’Sullivan, S.S.; Massey, L.A.; Williams, D.R.; Silveira-Moriyama, L.; Kempster, P.A.; Holton, J.L.; Revesz, T.; Lees, A.J. Clinical outcomes of progressive supranuclear palsy and multiple system atrophy. Brain 2008, 131, 1362–1372.
- Watanabe, H.; Saito, Y.; Terao, S.; Ando, T.; Kachi, T.; Mukai, E.; Aiba, I.; Abe, Y.; Tamakoshi, A.; Doyu, M.; et al. Progression and prognosis in multiple system atrophy: An analysis of 230 Japanese patients. Brain 2002, 125 Pt 5, 1070–1083.
- Marsili, L.; Rizzo, G.; Colosimo, C. Diagnostic Criteria for Parkinson’s Disease: From James Parkinson to the Concept of Prodromal Disease. Front. Neurol. 2018, 9, 156.
- Jellinger, K.A.; Logroscino, G.; Rizzo, G.; Copetti, M.; Arcuti, S.; Martino, D.; Fontana, A. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysisAuthor Response. Neurology 2016, 87, 237–238.
- Schrag, A.; Ben-Shlomo, Y.; Quinn, N. How valid is the clinical diagnosis of Parkinson’s disease in the community? J. Neurol. Neurosurg. Psychiatry 2002, 73, 529–534.
- Vaikath, N.N.; Hmila, I.; Gupta, V.; Erskine, D.; Ingelsson, M.; El-Agnaf, O.M.A. Antibodies against alpha-synuclein: Tools and therapies. J. Neurochem. 2019, 150, 612–625.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473.
- Hughes, A.J.; Colosimo, C.; Kleedorfer, B.; Daniel, S.E.; Lees, A.J. The dopaminergic response in multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 1992, 55, 1009–1013.
- Surguchov, A. Intracellular Dynamics of Synucleins: “Here, There and Everywhere”. Int. Rev. Cell Mol. Biol. 2015, 320, 103–169.
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425.
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841–842.
- Kay, D.M.; Factor, S.A.; Samii, A.; Higgins, D.S.; Griffith, A.; Roberts, J.W.; Leis, B.C.; Nutt, J.G.; Montimurro, J.S.; Keefe, R.G.; et al. Genetic association between α-synuclein and idiopathic parkinson’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 1222–1230.
- Ritz, B.; Rhodes, S.L.; Bordelon, Y.; Bronstein, J. α-Synuclein Genetic Variants Predict Faster Motor Symptom Progression in Idiopathic Parkinson Disease. PLoS ONE 2012, 7, e36199.
- Klos, K.J.; Ahlskog, J.E.; Josephs, K.A.; Apaydin, H.; Parisi, J.E.; Boeve, B.F.; DeLucia, M.W.; Dickson, D.W. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology 2006, 66, 1100–1102.
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67.
- Lee, P.H.; Shin, D.H.; Kim, J.W.; Song, Y.S.; Kim, H.S. Parkinsonism with basal ganglia lesions in a patient with uremia: Evidence of vasogenic edema. Park. Relat. Disord. 2006, 12, 93–96.
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240.
- Cersosimo, M.G. Gastrointestinal Biopsies for the Diagnosis of Alpha-Synuclein Pathology in Parkinson’s Disease. Gastroenterol. Res. Pract. 2015, 2015, 1–6.
- Cersosimo, M.G.; Perandones, C.; Micheli, F.E.; Raina, G.B.; Beron, A.M.; Nasswetter, G.; Radrizzani, M.; Benarroch, E.E. Alpha-synuclein immunoreactivity in minor salivary gland biopsies of Parkinson’s disease patients. Mov. Disord. 2011, 26, 188–190.
- Donadio, V.; Incensi, A.; Leta, V.; Giannoccaro, M.; Scaglione, C.; Cappellari, S.; Avoni, P.; Baruzzi, A.; Liguori, R. Skin nerve α-synuclein deposits: A biomarker for idiopathic Parkinson’s disease. Clin. Neurophysiol. 2015, 126, e19.
- Lebouvier, T.; Neunlist, M.; Des Varannes, S.B.; Coron, E.; Drouard, A.; N’Guyen, J.M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 2010, 5, e12728.
- Rodriguez-Leyva, I.; Chi-Ahumada, E.; Mejia, M.; Castanedo-Cazares, J.P.; Eng, W.; Saikaly, S.K.; Carrizales, J.; Levine, T.D.; Norman, R.A.; Jimenez-Capdeville, M.E. The Presence of Alpha-Synuclein in Skin from Melanoma and Patients with Parkinson’s Disease. Mov. Disord. Clin. Pract. 2017, 4, 724–732.
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nat. Cell Biol. 2011, 477, 107–110.
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of native brain α-synuclein. Nat. Cell Biol. 2013, 498, E4–E6.
- Fauvet, B.; Fares, M.B.; Samuel, F.; Dikiy, I.; Tandon, A.; Eliezer, D.; Lashuel, H.A. Characterization of semisynthetic and naturally Nalpha-acetylated alpha-synuclein in vitro and in intact cells: Implications for aggregation and cellular properties of alpha-synuclein. J. Biol. Chem. 2012, 287, 28243–28262.
- Levitan, K.; Chereau, D.; Cohen, S.I.A.; Knowles, T.P.J.; Dobson, C.M.; Fink, A.L.; Anderson, J.P.; Goldstein, J.M.; Millhauser, G.L. Conserved C-Terminal Charge Exerts a Profound Influence on the Aggregation Rate of α-Synuclein. J. Mol. Biol. 2011, 411, 329–333.
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283.
- Bellucci, A.; Zaltieri, M.; Navarria, L.; Grigoletto, J.; Missale, C.; Spano, P. From alpha-synuclein to synaptic dysfunctions: New insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012, 1476, 183–202.
- Deleersnijder, A.; Gerard, M.; Debyser, Z.; Baekelandt, V. The remarkable conformational plasticity of alpha-synuclein: Blessing or curse? Trends Mol. Med. 2013, 19, 368–377.
- Egallegos, S.; Epacheco, C.; Epeters, C.; Opazo, C.M.; Aguayo, L.G. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci. 2015, 9, 59.
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059.
- O’Hara, D.M.; Kalia, S.K.; Kalia, L.V. Methods for detecting toxic α-synuclein species as a biomarker for Parkinson’s disease. Crit. Rev. Clin. Lab. Sci. 2020, 57, 291–307.
- Dettmer, U.; Newman, A.J.; Von Saucken, V.E.; Bartels, T.; Selkoe, D.J. KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 9596–9601.
- Del Mar, C.; Greenbaum, E.A.; Mayne, L.; Englander, S.W.; Woods, V.L. Structure and properties of synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. USA 2005, 102, 15477–15482.
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073.
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Park. Dis. 2016, 6, 39–51.
- Wu, K.-P.; Kim, S.; Fela, D.A.; Baum, J. Characterization of Conformational and Dynamic Properties of Natively Unfolded Human and Mouse α-Synuclein Ensembles by NMR: Implication for Aggregation. J. Mol. Biol. 2008, 378, 1104–1115.
- Fernández, C.O.; Hoyer, W.; Zweckstetter, M.; Jares-Erijman, E.A.; Subramaniam, V.; Griesinger, C.; Jovin, T.M. NMR of α-synuclein–polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 2004, 23, 2039–2046.
- Giasson, B.I.; Lee, V.M.-Y.; Trojanowski, J.Q. Interactions of Amyloidogenic Proteins. Neuromolecular Med. 2003, 4, 49–58.
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Højrup, P.; Gliemann, J.; Jakes, R. α-Synuclein Binds to Tau and Stimulates the Protein Kinase A-catalyzed Tau Phosphorylation of Serine Residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489.
- Brown, D.R. Introduction: Copper and amyloid fibril formation. FEBS J. 2007, 274, 3755.
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the Acidic C-Terminal Region Comprising Amino Acids 109−140 on α-Synuclein Aggregation In Vitro. Biochemistry 2004, 43, 16233–16242.
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145.
- Machiya, Y.; Hara, S.; Arawaka, S.; Fukushima, S.; Sato, H.; Sakamoto, M.; Koyama, S.; Kato, T. Phosphorylated α-Synuclein at Ser-129 Is Targeted to the Proteasome Pathway in a Ubiquitin-independent Manner. J. Biol. Chem. 2010, 285, 40732–40744.
- Peng, C.; Gathagan, R.J.; Lee, V.M.-Y. Distinct α-Synuclein strains and implications for heterogeneity among α-Synucleinopathies. Neurobiol. Dis. 2018, 109, 209–218.
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103–117.
- Holec, S.A.M.; Woerman, A.L. Evidence of distinct α-synuclein strains underlying disease heterogeneity. Acta Neuropathol. 2020, 1–14.
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. α-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2019, 23, 21–31.
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nat. Cell Biol. 2020, 578, 273–277.
- Suzuki, G.; Imura, S.; Hosokawa, M.; Katsumata, R.; Nonaka, T.; Hisanaga, S.-I.; Saeki, Y.; Hasegawa, M. α-synuclein strains that cause distinct pathologies differentially inhibit proteasome. eLife 2020, 9, 9.