More emerging studies are exploring immunotherapy for solid cancers, including colorectal cancer. Besides, checkpoint blockade immunotherapy and chimeric antigen receptor (CAR) -based immune cell therapy have being examined in clinical trials for colorectal cancer patients. However, immunosuppression that leads to the blockage of normal immunosurveillance often leads to cancer development and relapse.
- Immunosuppression in Colorectal Cancer
1. Introduction
Colorectal cancer (CRC) is the second leading cause of cancer death worldwide, accounting for approximately 880,000 deaths in 2018[1]. Approximately 70–80% of patients with stage I–III CRC can be treated with curative surgical resection[2], and the 5-year survival rate for early-stage CRC is approximately 90%. However, the survival rate drops to only 10–15% for metastatic (stage IV) CRC[3]. Patients with stage IV CRC are mainly treated with fluorouracil-based chemotherapies, in combination with epidermal growth factor receptor (EGFR) or vascular endothelial growth factor (VEGF) inhibitors, and the median life expectancy is in the range of 21.3 to 30.1 months [4][5][6][7].
CRC is the most common gastrointestinal (GI) cancer. Compared with other tissues, the gastrointestinal tract is home to an enormous number of commensal bacteria and potential pathogens[8]. Therefore, the immune system in the colon and rectum has to fine-tune the balance of the maintenance of tolerance to self-tissue and immunity against potential pathogens and tumorigenic cells. Disruption of the GI tract’s normal immune homeostasis may cause autoimmunity diseases, including inflammatory bowel disease[9]. In contrast, normal immunosurveillance is critical to eliminate potential pathogens and eradicate sporadic tumorigenic cells to prevent GI diseases, including CRC.
It has been well recognized that the immune system plays a key role in colorectal cancer development and progression[10]. The presence of CD8
+ T cells in the tumor bed and invasive margin is strongly associated with disease-free survival and overall survival in CRC and the “immune score”, quantifying the density and location of immune cells within the tumor, has provided prognostic value that may be superior to the American Joint Committee on Cancer/Union for International Cancer Control (AJCC/UICC) tumor (T), nodes (N), and metastases (M) (TNM) classification in CRC[11][12]. This indicates that adaptive immune response may play a role in CRC progression[13] and immunotherapy may be promising in CRC treatment. However, the efficacy of immune checkpoint inhibitor monotherapy targeting programmed cell death-1 (PD-1) is only seen in a small subset of patients with microsatellite instability high (MSI-H) status. In contrast, no objective response was observed in microsatellite stable (MSS) CRCs, which account for the majority of the cases[14]. The mechanisms for resistance to anti-PD-1 immune checkpoint blockade in MSS CRC include lack of tumor neoantigens, immune exclusion, and immunosuppression. Higher tumor mutation burden (10- to 50-fold) and more neoantigens in MSI-H tumors, together with higher levels of multiple checkpoints, including PD-1, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), and lymphocyte activation gene 3 (LAG3) in MSI-H tumors, may explain in part why anti-PD-1 treatment works more efficiently in MSI-H CRCs than in MSS CRCs[15][16]. With the recent advancement in combination strategies and immunomodulatory monoclonal antibodies (mAbs) targeting other immune checkpoints, there is great enthusiasm in developing novel immunotherapies in CRC.
2. Current Immunotherapy Trials for MSS CRC
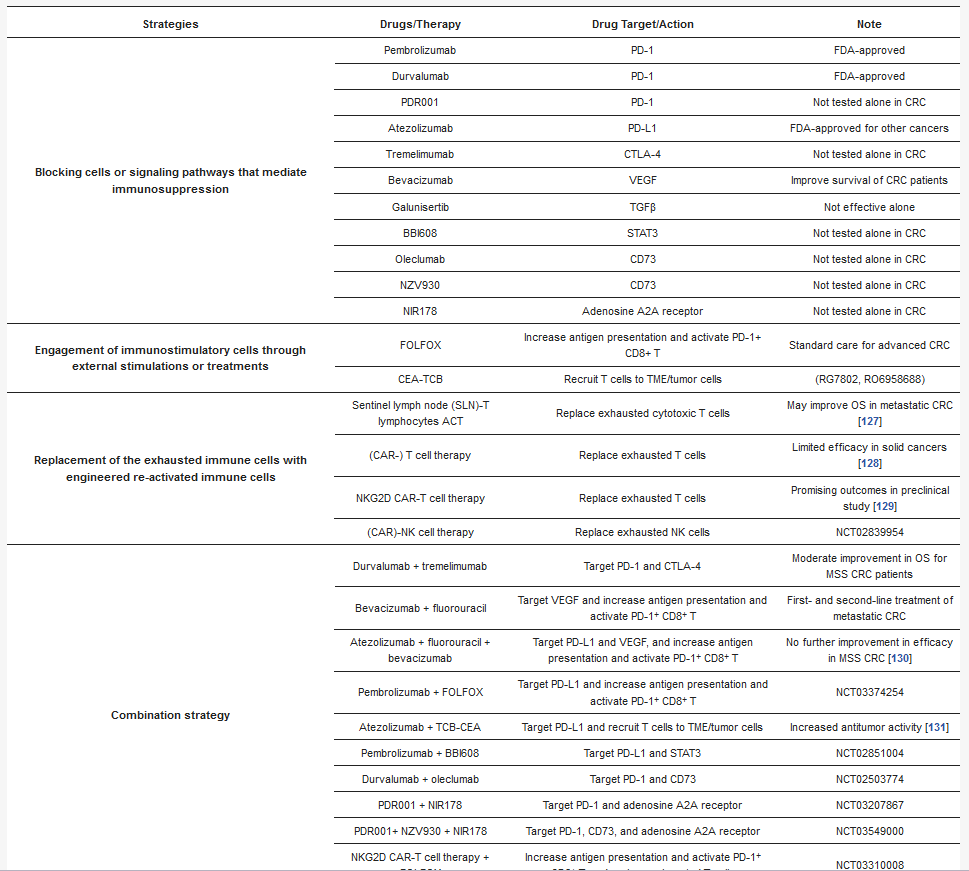
Table 1), including (1) suppression of the cells or signaling pathways that promote immunosuppression in TME; (2) engagement of immunostimulatory cells through external stimulations or treatments; and (3) replacement of the exhausted immune cells with engineered re-activated immune cells. Of course, the combination strategy may achieve the most promising result to deter immunosuppression in the TME.
| Strategies | Drugs/Therapy | Drug Target/Action | Note |
|---|---|---|---|
| Blocking cells or signaling pathways that mediate immunosuppression | Pembrolizumab | PD-1 | FDA-approved |
| Durvalumab | PD-1 | FDA-approved | |
| PDR001 | PD-1 | Not tested alone in CRC | |
| Atezolizumab | PD-L1 | FDA-approved for other cancers | |
| Tremelimumab | CTLA-4 | Not tested alone in CRC | |
| Bevacizumab | VEGF | Improve survival of CRC patients | |
| Galunisertib | TGFβ | Not effective alone | |
| BBI608 | STAT3 | Not tested alone in CRC | |
| Oleclumab | CD73 | Not tested alone in CRC | |
| NZV930 | CD73 | Not tested alone in CRC | |
| NIR178 | Adenosine A2A receptor | Not tested alone in CRC | |
| Engagement of immunostimulatory cells through external stimulations or treatments | FOLFOX | Increase antigen presentation and activate PD-1+ CD8+ T | Standard care for advanced CRC |
| CEA-TCB | Recruit T cells to TME/tumor cells | (RG7802, RO6958688) | |
| Replacement of the exhausted immune cells with engineered re-activated immune cells | Sentinel lymph node (SLN)-T lymphocytes ACT | Replace exhausted cytotoxic T cells | May improve OS in metastatic CRC [17] |
| (CAR-) T cell therapy | Replace exhausted T cells | Limited efficacy in solid cancers[18] | |
| NKG2D CAR-T cell therapy | Replace exhausted T cells | Promising outcomes in preclinical study[19] | |
| (CAR)-NK cell therapy | Replace exhausted NK cells | NCT02839954 | |
| Combination strategy | Durvalumab + tremelimumab | Target PD-1 and CTLA-4 | Moderate improvement in OS for MSS CRC patients |
| Bevacizumab + fluorouracil | Target VEGF and increase antigen presentation and activate PD-1+ CD8+ T | First- and second-line treatment of metastatic CRC | |
| Atezolizumab + fluorouracil + bevacizumab | Target PD-L1 and VEGF, and increase antigen presentation and activate PD-1+ CD8+ T | No further improvement in efficacy in MSS CRC [20] | |
| Pembrolizumab + FOLFOX | Target PD-L1 and increase antigen presentation and activate PD-1+ CD8+ T | NCT03374254 | |
| Atezolizumab + TCB-CEA | Target PD-L1 and recruit T cells to TME/tumor cells | Increased antitumor activity [21] | |
| Pembrolizumab + BBI608 | Target PD-L1 and STAT3 | NCT02851004 | |
| Durvalumab + oleclumab | Target PD-1 and CD73 | NCT02503774 | |
| PDR001 + NIR178 | Target PD-1 and adenosine A2A receptor | NCT03207867 | |
| PDR001+ NZV930 + NIR178 | Target PD-1, CD73, and adenosine A2A receptor | NCT03549000 | |
| NKG2D CAR-T cell therapy + FOLFOX | Increase antigen presentation and activate PD-1+ CD8+ T, and replace exhausted T cells | NCT03310008 |
Abbreviations: Folinic acid (leucovorin), Fluorouracil (5-FU), and Oxaliplatin (Eloxatin), FOLFOX; Carcinoembryonic Antigen T-Cell Bispecific antibody, CEA-TCB; adoptive cell transfer, ACT; overall survival, OS.
p
p = 0.07). There is no difference in progression-free survival (PFS) in the immunotherapy group compared to the best supportive care group[22].
VEGF-targeting therapies, such as bevacizumab, have been shown to attenuate the tumor-induced immunosuppressive microenvironment by decreasing the number of Tregs in both pre-clinical mouse models and patients with CRC[23][24]. However, adding atezolizumab (anti-PD-L1) to fluorouracil and bevacizumab as a first-line maintenance treatment for patients with metastatic MSS CRC did not result in improvement in efficacy in MSS CRC[20].
The alternative strategy is to block immunosuppressive cytokines that mediate the recruitment of Tregs, MDSCs, and TAMs. For example, TGF-β contributes to the immune exclusion in the TME at later stages of tumor development. Although TGF-β blockade as monotherapy is disappointing[25], the combination treatment of a small-molecule inhibitor (galunisertib) against TGF-β with anti-PD-1 or anti-PD-L1 agents has being tested in clinical trials of solid tumors (NCT02423343 and NCT02734160) and will likely yield promising outcomes. In addition, clinical trials with anti-PD1/PD-L1 in combination with anti-CD73, anti-adenosine A2A receptor, or triplet therapy are being examined in MSS CRC (NCT02503774, NCT03207867, and NCT03549000). The efficacy and safety of a STAT3 inhibitor (BBI608) in combination with pembrolizumab are also being assessed in a phase Ib/II study for patients with metastatic MSS CRC (NCT02851004).
+
+ T cells[26][27]. Therefore, immune checkpoint inhibitors (ICIs) in combination with standard-of-care chemotherapies may potentiate ICI efficacy by promoting a more immunogenic TME. In a phase II study, clinical activity was seen in the combination treatment of embrolizumab (anti-PD-1) with FOLFOX for patients with untreated advanced CRC, including those with proficient MMR (70%), even though the FOLFOX dose was reduced due to increased neutropenia in the initial cohort[28]. Besides, preliminary efficacy data showed a 70% objective response rate (ORR) for the combination treatment of pembrolizumab with FOLFOX in metastatic CRC patients with MMR-proficient disease in the first-line setting of an ongoing clinical trial (keynote-651 cohort B, NCT03374254)[29].
To target the immune-excluded TME in MSS CRC, a new strategy is to recruit T cells into the immunosuppressive TME by using T cell bispecific (TCB) antibodies. Bispecific antibody is designed to simultaneously bind two different epitopes or antigens, physically linking two binding specificities that may be temporally or spatially separate[30]. CEA-TCB (RG7802, RO6958688), a novel bispecific antibody that simultaneously binds to carcinoembryonic antigen (CEA) on tumor cells and CD3 on T cells, selectively engages effector T cells to kill CEA-expressing tumor cells. It was investigated in combination with atezolizumab in a phase 1 trial in patients with MSS CRC. Antitumor activity was observed during dose escalation with CEA-TCB monotherapy, with increased intratumoral CD3 T cell infiltration. Enhanced activity and a manageable safety profile were seen in combination with atezolizumab[21].
p = 0.02). This study showed that SLN-T lymphocyte immunotherapy was feasible and safe and may improve OS in metastatic CRC[17].
Chimeric antigen receptor (CAR)-T cell therapy uses chimeric T cell receptors encompassing an extracellular antigen-binding domain, transmembrane hinge domain, and an intracellular signaling domain with costimulatory molecules (such as CD28) in tandem with a CD3ζ chain. The CAR thereby reactivates the T cell immunity against tumor cells. However, CAR-T cell therapy has limited efficacy for the majority of solid tumors, due to limited trafficking and persistence of T cells into the tumor and an immunosuppressive TME[18]. Recently, a novel NKG2D CAR-T cell therapy, which uses a non-viral third-generation NKG2D CAR, yielded a promising outcome when tested for its antitumor activity against human CRC cells in vitro and in vivo[19]. To test the notion in a clinical trial, a phase I study is evaluating the safety and clinical activity of NKG2D CAR-T cells in combination with neoadjuvant FOLFOX in patients with metastatic CRC whose liver metastases are potentially resectable (NCT03310008). Additionally, NK cell-based cell therapy, which uses chimeric antigen receptor-expressing NK cells from the patient for anticancer therapy, has also made it to clinical trials for solid tumors, including CRC (NCT02839954)[31][32]. Further investigations will provide a more definitive conclusion if CAR-T cell therapies can benefit patients with MSS CRC.