Relapse after apparent remission remains a major cause of death in patients with acute myeloid leukemia (AML). On the cellular level, leukemia relapse is considered to emerge from subpopulations of therapy-resistant leukemic stem cells (LSC). Identification and targeting of LSC are thus most important goals for AML treatment. However, AML and their LSC are highly heterogeneous.
- Acute Myeloid Leukemia,Stem Cells
- heterogeneity
- relapse
1. Introduction
Acute myeloid leukemia (AML) is a devastating, rapidly-evolving disease characterized by an abnormal proliferation of poorly-differentiated cells which impairs normal hematopoiesis. AML patients suffer from cytopenia associated with recurrent infections, anemia, easy bleeding, and other manifestations [1] and show highly variable responses to therapy and survival rates. Notably, a major cause of disease progression and relapse is the persistence of therapy-resistant, clonogenic leukemic subpopulations: the leukemic stem cells (LSC) [2].
In 1994, John Dick and colleagues were the first to prove the existence of human LSC in an in vivo experimental model. Human CD34+ leukemic cells were shown to repopulate the bone marrow (BM) of severe combined immunodeficient (SCID) mice, while CD34− leukemic blasts remained non-leukemogenic [3][4]. These CD34+ cells responsible for leukemia initiation and maintenance were termed LSC. Nowadays, they are documented as cells with enhanced capacities to selectively escape chemotherapy treatments [5] as well as immune surveillance [6], thus leading to disease relapse after therapy, a major cause of death in these patients. Since AML is highly heterogeneous with respect to genetic alterations, epigenetics, and leukemia cell of origin, it is not surprising that considerable heterogeneity is also observed among surface markers of AML cells and their LSC [2], making immunological targeting of LSC a constant challenge [7][8][9].
Hematopoiesis is organized hierarchically with a minor subset of hematopoietic stem cells (HSC) giving rise to all blood cells during the lifespan of an individual. HSC must balance regenerative requirements (which naturally involve cell division and differentiation) with the need to protect their own genomic integrity by reducing cell division. In order to achieve this, HSC undergo highly complex fine-tuned interactions with the BM microenvironment and interact with several other cell types (e.g., osteoblasts, stromal cells, endothelial cells, adipocytes, and neural cells) via soluble factors, biophysical forces, and cell-mediated interactions [10]. Similarly, LSC also reside and are influenced by the so-called BM niche, which sustains their quiescence and protects them from genotoxic stress [11][12].
AML is also organized hierarchically and contains subpopulations of LSC that share functional and molecular properties with their cells of origin, the healthy hematopoietic stem and progenitor cells (HSPC) [4][13][14][15]. Consistent with a close relationship between these two cell types, molecules expressed on healthy HSPC, i.e., CD34, were also reported to identify LSC [16]. Functionally, the CD34 family encompasses podocalyxin and endoglycan proteins and is considered to regulate cell differentiation, adhesion, trafficking, and proliferation [17]. CD34 is expressed on the vast majority of HSC, but rare CD34− HSC giving rise to CD34+ HSPC have also been reported [16].
In 2016, the LSC17 gene expression score was defined as the molecular LSC hallmark that predicts outcome and treatment resistance in patients with AML [18]. Among the genes highlighted in the LSC17 score were e.g., CD34 and the G protein-coupled receptor GPR56, a surface protein involved in cell adhesion which was also described to mark healthy HSC [16][19]. However, great phenotypic heterogeneity is observed in AML LSC and a wide range of surface markers has been found to identify LSC in only some, but not all AML (e.g., CD93, TIM3, CD44, CD123, etc. [9][20][21][22][23][24][25][26]).
2. The Relevance of Immunomodulatory Proteins for LSC Detection
Interestingly, a variety of antigens involved in LSC identification are in fact involved in immunological processes (Figure 1, Table 1). This suggests that LSC and non-LSC may have different interactions with the immune system. This notion has been substantiated by recent work from our research group demonstrating that LSC selectively escape immune surveillance by suppressing surface expression of NKG2D ligands (NKG2DL) [6]. When compared to corresponding non-stem leukemic blasts from the same patients, LSC lack expression of such ligands for activating NKG2D receptors on natural killer (NK) cells thereby evading NK-mediated lysis. In several AML patient samples of heterogeneous genetic backgrounds, lack of NKG2DL surface expression robustly distinguished LSC from other non-stem leukemic cells [6].
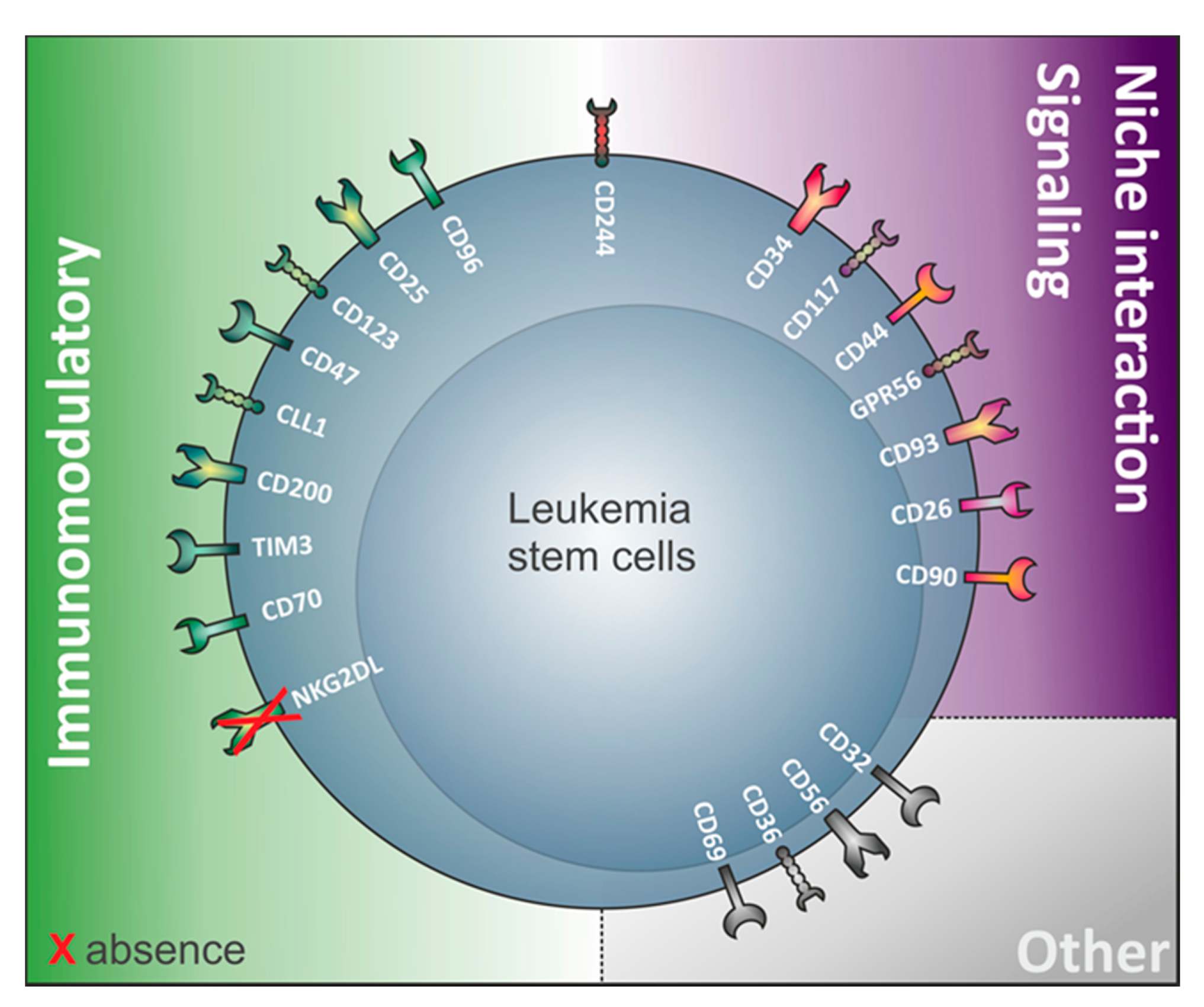
Table 1. Non-comprehensive list of human markers that can be found on LSC and their (potential) expression on the cell surface of other healthy blood cells. Highlighted in grey are the markers expressed on the cell surface of LSC from both CD34-expressing and non-expressing acute myeloid leukemia (AML). In white: markers only demonstrated to play roles in LSC from CD34 expressing AML. N.D: Not described/MPP: multipotential progenitor/MEP: megakaryocyte–erythroid progenitor).
| Antigen | Percentage of AML Patients Expressing the Marker | Expression on Non-LSC | Expression on HSC | Expression on Other Healthy Blood Cells | Function in Healthy Conditions | References |
|---|---|---|---|---|---|---|
| CLL-1 | 92 | Yes | No | Monocytes, granulocytes, CMP, GMP | Modulates the activation state of cells during inflammation processes | Bakker et al. 2004 [27] Jiang et al. 2018 [28] Daga et al. 2019 [29] Marshall et al. 2006 [30] |
| CD9 | 40 | Yes | No | Monocytes, macrophages, granulocytes, DC, endothelial cells, B, T, and NK cells | Cell migration, adhesion, activation, | Brosseau et al. 2018 [31] Touzet et al. 2019 [32] Paprocka et al. 2017 [33] |
| CD25 | 10–25 | Yes | No | T cells and regulatory T cells | Important role for T cells survival | Saito et al. 2010 [34] Kageyama et al. 2018 [35] Triplett et al. 2012 [36] |
| CD26 | N.D | Yes | No | T, B, NK, and myeloid cells | T cell activation and proliferation, cell adhesion, metabolism | Herrmann et al. 2020 [25] Klemann et al. 2016 [37] |
| CD32 | 35 | Yes | No | Monocytes, B and T cells | Immune cell activation | Saito et al. 2010 [34] Anania et al. 2019 [38] |
| CD33 | 88 | Yes | Yes | Myeloid cells, lymphocytes, NK cells, MPP, GMP, MEP | Modulates inflammatory and immune responses by reducing tyrosine kinase dependent pathways | Ehninger et al. 2014 [39] Liu et al. 2007 [40] Laszlo et al. 2014 [41] Haubner et al. 2017 [24] |
| CD34 | 70 | Yes | Yes | Mast cells, eosinophils, neurons, fibrocytes | Regulates cell differentiation, adhesion, trafficking and proliferation | Quek et al. 2016 [42] Engelhardt et al. 2002 [16] Nielsen et al. 2008 [17] |
| CD36 | N.D | Yes | No | Platelets, monocytes, adipocytes | Fatty acid uptake, angiogenesis, PRR recognition | Silverstein et al. 2009 [43] Sachs et al. 2020 [44] Herrmann et al. 2020 [25] |
| CD38 | 5–55 (FAB subtypes) |
Yes | No | T and B cells, monocytes, NK, granulocytes, platelets, red blood cells | Regulates calcium levels and NAD+ homeostasis | Hogan et al. 2019 [45] Sarry et al. 2011 [46] Goardon et al. 2011 [47] Keyhani et al. 2000 [48] |
| CD44 | N.D | Yes | Yes | T cells, mesenchymal cells, ectodermal cells, neuron-like cells | Cell adhesion molecule, cellular signaling | Ponta et al. 2003 [49] Jin et al. 2006 [50] Bendall et al. 2000 [51] Herrmann et al. 2020 [25] |
| CD45RA | N.D | Yes | Yes | T and B cells | CD45 isoform, cell signaling | Kersten et al. 2016 [52] Goardon et al. 2011 [47] Sarry et al. 2011 [46] Holmes 2006 [53] |
| CD47 | N.D | Yes | Yes | Various healthy cells | “don’t eat me” signal on cells in order to prevent inappropriate phagocytosis | Majeti et al. 2009 [54] Jaiswal et al. 2009 [55] Sick et al. 2012 [56] |
| CD56 | Up to 20 | Yes | No | DC, T and NK cells | Linked to NK cytotoxicity | Van Acker et al. 2017 [57] Sasca et al. 2019 [58] Herrmann et al. 2020 [25] |
| CD69 | N.D | N.D | No | T cells | T cell differentiation, tissue retention, and metabolic reprogramming | Cibrián et al. 2017 [59] Sachs et al. 2020 [44] Herrmann et al. 2020 [25] |
| CD70 | N.D | Yes | No | DC | T and B cell activation | Riether et al. 2015 [60] Riether et al. 2017 [61] Borst et al. 2005 [62] |
| CD90 | 40 (in elderly patients) | Yes | Yes | Fibroblasts, neurons, endothelial cells | Maintenance of HSC, cell adhesion, matrix adhesion | Buccisano et al. 2004 [63] Blair et al. 1997 [64] Brendel et al. 1999 [65] Kisselbach et al. 2009 [66] Craig et al. 1993 [67] |
| CD93 | N.D | N.D | No (only on CD34-HSC) | Myeloid and endothelial cells | Mechanism in innate host defense | Bohlson et al. 2008 [68] Iwasaki et al. 2015 [69] Sumide et al. 2018 [70] |
| CD96 | 27 | Yes | Only 5% | T and NK cells | Inhibits NK and T cells | Fatlawi et al. 2016 [71] Georgiev et al. 2018 [72] Hosen et al. 2007 [73] |
| CD117 | 87 | Yes | Yes | GMP | Promotes HSC growth by binding the stem cell factor | Sperling et al. 1997 [74] Geissler et al. 1991 [75] Quek et al. 2016 [42] Wells et al. 1996 [76] |
| CD123 | 97 | Yes | No | Basophils, plasmacytoid DC | Proliferation, survival, activation, and differentiation by binding respective ligand | Yu et al. 2016 [76] Guthridge et al. 1998 [77] Bras et al. 2019 [78] Haubner et al. 2019 [24] Al-Mawali et al. 2017 [79] |
| CD200 | N.D | Yes | Yes | Myeloid, T and B cells | Immunoregulatory molecule | Ngwa et al. 2019 [80] Ho et al. 2020 [81] |
| CD244 | N.D | Yes | Yes | GMP, HSPC, granulocytes, monocytes, DC, NK and T cells | Regulates NK, T, and DC activation state | Zhang et al. 2017 [82] Haubner et al. 2019 [24] Quek et al. 2016 [42] Agresta et al. 2018 [83] |
| GPR56 | N.D | No | Yes | Central nervous system, T cells | Frontal cortex development, NK inhibition, cell migration, HSC generation | Pabst et al. 2016 [84] Daga et al. 2019 [29] Kartalaei et al. 2015 [85] Huang et al. 2018 [86] |
| NKG2DL (its absence defines LSC) |
Highly variable | Yes | No | Not expressed on healthy cells | Upregulation of NG2DL on malignant or virus-infected cells resulting in their clearance by NK cells | Paczulla et al. 2019 [6] Zingoni et al. 2018 [87] |
| TIM-3 | 98 | Yes | No | T cells, monocytes, macrophages, DC, and mast cells | Homeostasis-maintaining molecule of the immune system | Jan et al. 2011 [88] Haubner et al. 2019 [24] Kikushige et al. 2010 [89] Han et al. 2013 [90] |
Other examples of immunomodulatory proteins involved in LSC identification (Figure 1) include the immunoglobulin superfamily member CD96, a molecule expressed on healthy T and natural killer cells with known inhibitory roles on NK cells [72], TIM-3 (T cell immunoglobulin mucin-3), a homeostasis-maintaining molecule of the immune system expressed on the surface of CD4+ T type 1 helper cells (Th1) and CD8+ T type 1 cytotoxic cells, monocytes/macrophages, dendritic cells (DC), and mast cells [90], the lectin protein CLL-1 regulating cell activation during inflammation and CD32, an immune-activating immunoglobulin Fc receptor family member showing broad expression on hematopoietic cells [30][38]. Furthermore, the interleukin-2 receptor alpha-chain CD25 commonly expressed on activated and regulatory T cells, but also found on resting memory T cells [36], and CD123, the interleukin-3 receptor (IL-3R) alpha chain, which is part of the IL-3R system that includes interleukin-5 receptor (IL-5R) and granulocyte-macrophage colony stimulating factor receptor (GM-CSFR), are also found on LSC. While interleukin 2 is important for survival, activation, and proliferation of T cells, the IL-3R system influences proliferation, survival, and differentiation of hematopoietic cells and is involved in immunity and inflammatory response by specifically binding respective ligands (IL-3, IL-5, and GM-CSF) [77].
Last but not least, the immunoglobulin-like and integrin-associated protein CD47 was identified as a novel AML LSC marker [55]. CD47 serves as a ligand of signal regulatory protein-1 (SIRP-1) and thereby functions as a “don’t eat me” signal, protecting LSC from macrophage phagocytosis [54].
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737.
- Bahr, C.; Correia, N.C.; Trumpp, A. Stem cells make leukemia grow again. EMBO J. 2017, 36, 2667–2669.
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259.
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678.
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635.
- Wang, X.; Huang, S.; Chen, J.L. Understanding of leukemic stem cells and their clinical implications. Mol. Cancer 2017, 16, 2.
- Guilak, F.; Cohen, D.M.; Estes, B.T.; Gimble, J.M.; Liedtke, W.; Chen, C.S. Control of Stem Cell Fate by Physical Interactions with the Extracellular Matrix. Cell Stem Cell 2009, 5, 17–26.
- Hoggatt, J.; Kfoury, Y.; Scadden, D.T. Hematopoietic Stem Cell Niche in Health and Disease. Annu. Rev. Pathol. 2016, 11, 555–581.
- Sánchez-Aguilera, A.; Méndez-Ferrer, S. The hematopoietic stem-cell niche in health and leukemia. Cell. Mol. Life Sci. 2017, 74, 579–590.
- Passegué, E.; Jamieson, C.H.M.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849.
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093.
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333.
- Engelhardt, M.; Lübbert, M.; Guo, Y. CD34+ or CD34−: Which is the more primitive? Leukemia 2002, 16, 1603–1608.
- Nielsen, J.S.; McNagny, K.M. Erratum: Novel functions of the CD34 family. J. Cell Sci. 2008, 121, 3683–3692.
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437.
- Saito, Y.; Morishita, K. Maintenance of leukemic and normal hematopoietic stem cells in bone marrow niches by EVI1-regulated GPR56. Rinsho Ketsueki 2015, 56, 375–383.
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557.
- Zagozdzon, R.; Golab, J. Cancer stem cells in haematological malignancies. Wspolczesna Onkol. 2015, 19, A1–A6.
- Zeijlemaker, W.; Kelder, A.; Cloos, J.; Schuurhuis, G.J. Immunophenotypic Detection of Measurable Residual (Stem Cell) Disease Using LAIP Approach in Acute Myeloid Leukemia. Curr. Protoc. Cytom. 2019, 91, e66.
- Zhou, J. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J. Stem Cells 2014, 6, 473–484.
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74.
- Herrmann, H.; Sadovnik, I.; Eisenwort, G.; Thomas, R.; Blatt, K.; Herndlhofer, S.; Willmann, M.; Stefanzl, G.; Baumgartner, S.; Greiner, G.; et al. Delineation of target expression profiles in CD34+/CD38− and CD34+/CD38− stem and progenitor cells in AML and CML. Blood Adv. 2020, 4, 5118–5132.
- Zeijlemaker, W.; Kelder, A.; Oussoren-Brockhoff, Y.J.M.; Scholten, W.J.; Snel, A.N.; Veldhuizen, D.; Cloos, J.; Ossenkoppele, G.J.; Schuurhuis, G.J. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia 2016, 30, 439–446.
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.; Visser, T.J.; Bijl, N.; Geuijen, C.A.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450.
- Jiang, Y.P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749.
- Daga, S.; Rosenberger, A.; Quehenberger, F.; Krisper, N.; Prietl, B.; Reinisch, A.; Zebisch, A.; Sill, H.; Wolfler, A. High GPR56 surface expression correlates with a leukemic stem cell gene signature in CD34-positive AML. Cancer Med. 2019, 8, 1771–1778.
- Marshall, A.S.J.; Willment, J.A.; Pyz, E.; Dennehy, K.M.; Reid, D.M.; Dri, P.; Gordon, S.; Wong, S.Y.C.; Brown, G.D. Human MICL (CLEC12A) is differentially glycosylated and is down-regulated following cellular activation. Eur. J. Immunol. 2006, 36, 2159–2169.
- Brosseau, C.; Colas, L.; Magnan, A.; Brouard, S. CD9 tetraspanin: A new pathway for the regulation of inflammation? Front. Immunol. 2018, 9, 2316.
- Touzet, L.; Dumezy, F.; Roumier, C.; Berthon, C.; Bories, C.; Quesnel, B.; Preudhomme, C.; Boyer, T. CD9 in acute myeloid leukemia: Prognostic role and usefulness to target leukemic stem cells. Cancer Med. 2019, 8, 1279–1288.
- Paprocka, M.; Bielawska-Pohl, A.; Rossowska, J.; Krawczenko, A.; Duś, D.; Kiełbiński, M.; Haus, O.; Podolak-Dawidziak, M.; Kuliczkowski, K. MRP1 protein expression in leukemic stem cells as a negative prognostic marker in acute myeloid leukemia patients. Eur. J. Haematol. 2017, 99, 415–422.
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci. Transl. Med. 2010, 2, 17ra19.
- Kageyama, Y.; Miwa, H.; Arakawa, R.; Tawara, I.; Ohishi, K.; Masuya, M.; Nakase, K.; Katayama, N. Expression of CD25 fluctuates in the leukemia-initiating cell population of CD25-positive AML. PLoS ONE 2018, 13, e0209295.
- Triplett, T.A.; Curti, B.D.; Bonafede, P.R.; Miller, W.L.; Walker, E.B.; Weinberg, A.D. Defining a functionally distinct subset of human memory CD4+ T cells that are CD25POS and FOXP3NEG. Eur. J. Immunol. 2012, 42, 1893–1905.
- Klemann, C.; Wagner, L.; Stephan, M.; von Hörsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21.
- Anania, J.C.; Chenoweth, A.M.; Wines, B.D.; MarkHogarth, P. The human FcγRII (CD32) family of leukocyte FCR in health and disease. Front. Immunol. 2019, 10, 464.
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014, 4, e218.
- Liu, X.-L.; Yuan, J.-Y.; Zhang, J.-W.; Zhang, X.-H.; Wang, R.-X. Differential gene expression in human hematopoietic stem cells specified toward erythroid, megakaryocytic, and granulocytic lineage. J. Leukoc. Biol. 2007, 82, 986–1002.
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153.
- Quek, L.; Otto, G.W.; Garnett, C.; Lhermitte, L.; Karamitros, D.; Stoilova, B.; Lau, I.J.; Doondeea, J.; Usukhbayar, B.; Kennedy, A.; et al. Genetically distinct leukemic stem cells in human CD34− acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J. Exp. Med. 2016, 213, 1513–1535.
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3.
- Sachs, K.; Sarver, A.L.; Noble-Orcutt, K.E.; LaRue, R.S.; Antony, M.L.; Chang, D.; Lee, Y.; Navis, C.M.; Hillesheim, A.L.; Nykaza, I.R.; et al. Single-cell gene expression analyses reveal distinct self-renewing and proliferating subsets in the leukemia stem cell compartment in acute myeloid leukemia. Cancer Res. 2020, 80, 458–470.
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 2019, 10, 1187.
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Recher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J. Clin. Investig. 2011, 121, 384–395.
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152.
- Keyhani, A.; Huh, Y.O.; Jendiroba, D.; Pagliaro, L.; Cortez, J.; Pierce, S.; Pearlman, M.; Estey, E.; Kantarjian, H.; Freireich, E.J. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk. Res. 2000, 24, 153–159.
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45.
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12, 1167–1174.
- Bendall, L.J.; Bradstock, K.F.; Gottlieb, D.J. Expression of CD44 variant exons in acute myeloid leukemia is more common and more complex than that observed in normal blood, bone marrow or CD34+ cells. Leukemia 2000, 14, 1239–1246.
- Kersten, B.; Valkering, M.; Wouters, R.; van Amerongen, R.; Hanekamp, D.; Kwidama, Z.; Valk, P.; Ossenkoppele, G.; Zeijlemaker, W.; Kaspers, G.; et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br. J. Haematol. 2016, 173, 219–235.
- Holmes, N. CD45: All is not yet crystal clear. Immunology 2006, 117, 145–155.
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299.
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285.
- Sick, E.; Jeanne, A.; Schneider, C.; Dedieu, S.; Takeda, K.; Martiny, L. CD47 update: A multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 2012, 167, 1415–1430.
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the immune system: More than a marker for cytotoxicity? Front. Immunol. 2017, 8, 892.
- Sasca, D.; Szybinski, J.; Schüler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.M.; Gebhardt, W.H.; et al. NCAM1 (CD56) promotes leukemogenesis and confers drug resistance in AML. Blood 2019, 133, 2305–2319.
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953.
- Riether, C.; Schürch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187–198.
- Riether, C.; Schürch, C.M.; Bührer, E.D.; Hinterbrandner, M.; Huguenin, A.L.; Hoepner, S.; Zlobec, I.; Pabst, T.; Radpour, R.; Ochsenbein, A.F. CD70/CD27 signaling promotes blast stemness and is a viable therapeutic target in acute myeloid leukemia. J. Exp. Med. 2017, 214, 359–380.
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005, 17, 275–281.
- Buccisano, F.; Rossi, F.M.; Venditti, A.; Del Poeta, G.; Cox, M.C.; Abbruzzese, E.; Rupolo, M.; Berretta, M.; Degan, M.; Russo, S.; et al. CD90/Thy-1 is preferentially expressed on blast cells of high risk acute myeloid leukaemias. Br. J. Haematol. 2004, 125, 203–212.
- Blair, A.; Hogge, D.E.; Ailles, L.E.; Lansdorp, P.M.; Sutherland, H.J. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 1997, 89, 3104–3112.
- Brendel, C.; Mohr, B.; Schimmelpfennig, C.; Muller, J.; Bornhauser, M.; Schmidt, M.; Ritter, M.; Ehninger, G.; Neubauer, A. Detection of cytogenetic aberrations both in CD90 (Thy-1)-positive and (Thy-1)-negative stem cell (CD34) subfractions of patients with acute and chronic myeloid leukemias. Leukemia 1999, 13, 1770–1775.
- Kisselbach, L.; Merges, M.; Bossie, A.; Boyd, A. CD90 expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology 2009, 59, 31–44.
- Craig, W.; Kay, R.; Cutler, R.L.; Lansdorp, P.M. Expression of Thy-1 on human hematopoietic progenitor cells. J. Exp. Med. 1993, 177, 1331–1342.
- Bohlson, S.; Greenlee, M.; Sullivan, S. CD93 and Related Family Members: Their Role in Innate Immunity. Curr. Drug Targets 2008, 9, 130–138.
- Iwasaki, M.; Liedtke, M.; Gentles, A.J.; Cleary, M.L. CD93 marks a non-quiescent human leukemia stem cell population and is required for development of MLL-rearranged acute myeloid leukemia. Cell Stem Cell 2015, 17, 412–421.
- Sumide, K.; Matsuoka, Y.; Kawamura, H.; Nakatsuka, R.; Fujioka, T.; Asano, H.; Takihara, Y.; Sonoda, Y. A revised road map for the commitment of human cord blood CD34-negative hematopoietic stem cells. Nat. Commun. 2018, 9, 2202.
- Al-Fatlawi, H.; Musa, R. Evaluation of CD96 and CD123 in CD34+ leukemic stem cells in acute myeloid leukemia patients and their relation to response to induction therapy. Iraqi J. Hematol. 2016, 5, 161–166.
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of age: CD96 emerges as modulator of immune responses. Front. Immunol. 2018, 9, 1072.
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 11008–11013.
- Sperling, C.; Schwartz, S.; Büchner, T.; Thiel, E.; Ludwig, W.D. Expression of the stem cell factor receptor C-KIT (CD117) in acute leukemias. Haematologica 1997, 82, 617–621.
- Geissler, E.N.; Liao, M.; Brook, J.D.; Martin, F.H.; Zsebo, K.M.; Housman, D.E.; Galli, S.J. Stem cell factor (SCF), a novel hematopoietic growth factor and ligand for c-kit tyrosine kinase receptor, maps on human chromosome 12 between 12q14.3 and 12qter. Somat Cell Mol. Genet. 1991, 17, 207–214.
- Wells, S.J.; Bray, R.A.; Stempora, L.L.; Farhi, D.C. CD117/CD34 expression in leukemic blasts. Am. J. Clin. Pathol. 1996, 106, 192–195.
- Guthridge, M.A.; Stomski, F.C.; Thomas, D.; Woodcock, J.M.; Bagley, C.J.; Berndt, M.C.; Lopez, A.F. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells 1998, 16, 301–313.
- Bras, A.E.; de Haas, V.; van Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; te Marvelde, J.G.; Zwaan, C.M.; van Dongen, J.J.M.; Leusen, J.H.W.; van der Velden, V.H.J. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytom. Part B Clin. Cytom. 2019, 96, 134–142.
- Al-Mawali, A.; Pinto, A.D.; Al-Zadjali, S. CD34+CD38-CD123+ Cells Are Present in Virtually All Acute Myeloid Leukaemia Blasts: A Promising Single Unique Phenotype for Minimal Residual Disease Detection. Acta Haematol. 2017, 138, 175–181.
- Ngwa, C.; Liu, F. CD200-CD200R signaling and diseases: A potential therapeutic target? Int. J. Physiol. Pathophysiol. Pharmcol. 2019, 11, 297–309.
- Ho, J.M.; Dobson, S.M.; Voisin, V.; McLeod, J.; Kennedy, J.A.; Mitchell, A.; Jin, L.; Eppert, K.; Bader, G.; Minden, M.D.; et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020, 4, 5402–5413.
- Zhang, F.; Liu, X.; Chen, C.; Zhu, J.; Yu, Z.; Xie, J.; Xie, L.; Bai, H.; Zhang, Y.; Fang, X.; et al. CD244 maintains the proliferation ability of leukemia initiating cells through SHP-2/p27kip1 signaling. Haematologica 2017, 102, 707–718.
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front. Immunol. 2018, 9, 2809.
- Pabst, C.; Bergeron, A.; Lavallée, V.P.; Yeh, J.; Gendron, P.; Norddahl, G.L.; Krosl, J.; Boivin, I.; Deneault, E.; Simard, J.; et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016, 127, 2018–2027.
- Solaimani Kartalaei, P.; Yamada-Inagawa, T.; Vink, C.S.; de Pater, E.; van der Linden, R.; Marks-Bluth, J.; van der Sloot, A.; van den Hout, M.; Yokomizo, T.; van Schaick-Solerno, M.L.; et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J. Exp. Med. 2015, 212, 93–106.
- Huang, K.Y.; Lin, H.H. The activation and signaling mechanisms of GPR56/ADGRG1 in melanoma cell. Front. Oncol. 2018, 8, 304.
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and its ligands: “One for all, all for one”. Front. Immunol. 2018, 9, 476.
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014.
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717.
- Han, G.; Chen, G.; Shen, B.; Li, Y. Tim-3: An activation marker and activation limiter of innate immune cells. Front. Immunol. 2013, 4, 449.
- Zhang, F.; Liu, X.; Chen, C.; Zhu, J.; Yu, Z.; Xie, J.; Xie, L.; Bai, H.; Zhang, Y.; Fang, X.; et al. CD244 maintains the proliferation ability of leukemia initiating cells through SHP-2/p27kip1 signaling. Haematologica 2017, 102, 707–718.
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front. Immunol. 2018, 9, 2809.
- Pabst, C.; Bergeron, A.; Lavallée, V.P.; Yeh, J.; Gendron, P.; Norddahl, G.L.; Krosl, J.; Boivin, I.; Deneault, E.; Simard, J.; et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016, 127, 2018–2027.
- Solaimani Kartalaei, P.; Yamada-Inagawa, T.; Vink, C.S.; de Pater, E.; van der Linden, R.; Marks-Bluth, J.; van der Sloot, A.; van den Hout, M.; Yokomizo, T.; van Schaick-Solerno, M.L.; et al. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J. Exp. Med. 2015, 212, 93–106.
- Huang, K.Y.; Lin, H.H. The activation and signaling mechanisms of GPR56/ADGRG1 in melanoma cell. Front. Oncol. 2018, 8, 304.
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and its ligands: “One for all, all for one”. Front. Immunol. 2018, 9, 476.
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc. Natl. Acad. Sci. USA 2011, 108, 5009–5014.
- Kikushige, Y.; Shima, T.; Takayanagi, S.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y.; et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 2010, 7, 708–717.