Proteins are localized into different cellular compartments and sub-compartments inside the cell. Each subcellular compartment has a distinct well-defined function in the cell and has a characteristic physicochemical environment, which drives proper functioning of the proteins. Each subcellular compartment has a distinct, well defined function in the cell and is considered to have evolved from the prokaryotic cell. Typical eukaryotic cells have two types of DNAs (i) chromosomal nuclear DNA and (ii) organelle DNA, which is present in mitochondria and chloroplast while prokaryotic cells have only single type of DNA called nucleoid.
- protein localization
1. Introduction
The nuclear DNA encodes the majority of proteins while only a small number of proteins are encoded by organelle DNA. Eukaryotic cells can synthesize up to 100,000 different types of protein [1], which are destined for one or more predetermined subcellular locations. Figure 1 depicts various protein localization prediction methods available for different cellular compartments.
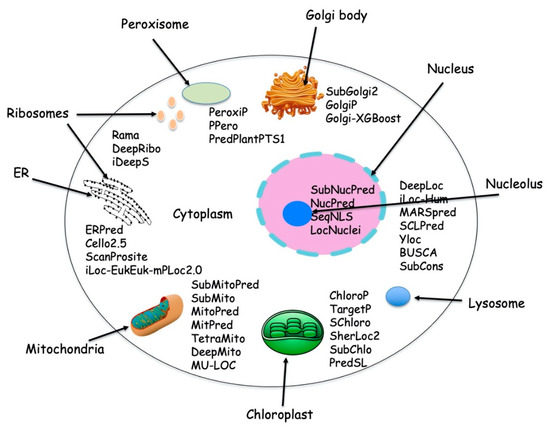
Figure 1. Typical cell with different subcellular location and with available protein localization prediction tools.
The protein synthesis occurs in the cytoplasm and then the newly synthesized proteins are further transported to their destined compartment to execute their function. Protein must be targeted to the right compartment in cells to perform their function and mis-localization of the proteins leads to functional loss or disorder, which contributes to many human diseases including cardiovascular, neurodegenerative disease and cancers [2][3][2,3]. Assigning subcellular localization for protein is a significant step to elucidate its interaction partners and predict their functions or potential roles in the cellular machinery [4]. There are a number of sequences that are deposited every year in the UniProt Knowledgebase (UniProtKB) but only a few of them were manually annotated and reviewed (UniProtKB/SwissProt), which explains the gap between the deposited sequence and annotated sequence is increasing every year (Figure 2). Therefore, there is a need of computational methods to predict subcellular localization with high quality and accuracy, which is of great significance in understanding cellular proteome and also helpful in designing the drug or targets. To date, many efforts have been made in this regard. Based on different kinds of characteristics, several machine learning approaches have been developed such as neural networks [5][6][5,6], hidden Markov models [7][8][9][7,8,9], support vector machines [10][11][12][10,11,12], deep learning [13][14][15][13,14,15], random forest [16], and extreme gradient boosting [17] [17] for prediction of subcellular localization of proteins.
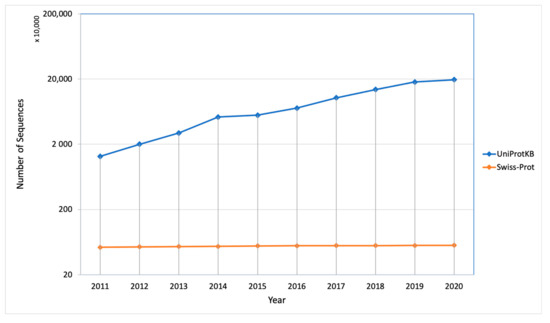
Figure 2. Number of sequences deposited and manually annotated proteins in the UniProt database in the last 10 years.
2. Experimental Approaches for Protein Localization
Several experimental methods are available for determining protein localization, but the most common method is to label the protein of interest with fluorescent probes and then visualize the distribution of the protein within cells under a fluorescence microscope such as immunofluorescence microscopy, immunolocalization, mass spectrometry, co-expression of fluorescent proteins, and electron microscopy. Fractionation based approach such as gradient centrifugation and 2D gel electrophoresis are also a widely used method to experimentally establish the localization of a protein. These experimental methods are relatively expensive and time consuming, which explains for a large information gap existing between known protein and their location information. Consequently, various computational methods have been developed to help fill this void. In this review, we focused only on the computational approaches and tools for prediction of protein localization.
3. Computational Approaches for Protein Localization
With the rapid development of advanced genome sequencing methods, the complete genome sequences are increasing day by day and the challenges for computational biologists are to manage, analyze, and annotate this plethora of unprocessed raw biological data. To now, a number of computational methods have been developed to solve this problem. While many have attempted to explore uncharacterized protein information, others have used the whole proteome sequence information to develop new machine learning algorithms for different things such as the prediction of motifs, prediction of ligand binding sites, etc. Based on protein sequence information, the computational method can be divided into the following categories: (1) sequence feature-based methods, (2) homology-based methods, (3) protein domain and motif information-based methods, (4) signal peptide-based methods, (5) non-sequence derived features-based methods, and (6) integrated methods, which could use a combination of two or more methods.
3.1. Sequence Feature-Based Method
Sequence features are commonly used in localization prediction since some differences in the sequence features are empirically known to be correlated with different localization sites. Nishikawa and Ooi [18] [18] first noted the correlation of amino acid composition to its biological and functional character in 1982. After that in 1983 Nakashima developed the first sequence-based method for subcellular localization [19]. They used amino acid composition to discriminate between intracellular and extracellular proteins. Later several research groups successfully used amino acid composition as a tool for subcellular localization predictions [16][20][21][16,20,21].
In sequence feature-based methods, the complete sequence of proteins is transformed into a numerical feature vector, which is then used to predict the subcellular location. There are different types of sequence feature-based methods available: (i) amino acid composition based method, in which the frequency of 20 different amino acids is calculated but it ignores the sequence order information of each residue. (ii) Chou’s Pseudo amino acid composition (PseAAC) [22], which considers the amino acid composition along with the potential interaction among the adjacent residues. This can be further categorized into different modes of PseAAC such as the gene ontology mode, functional domain mode and sequential evolution mode. (iii) Hybrid method, which allows the integration of different parameters or features for the prediction and usually results in an increased the prediction performance [4].
3.2. Homology Based Method
This is the most common way to predict the uncharacterized protein on the basis of the presence of homologous sequences of known function with an assumption that function is evolutionarily conserved [23]. This approach first identifies for homologous sequences in the proteins with known subcellular location and then extrapolates to predict the location of unknown proteins, hence this approach is also known as “Annotation by Homology Transfer”. Homology is a qualitative term, which attributes evolutionary relationships among different protein sequences. Orthologous proteins also typically have similar sequences and thus similar subcellular localization patterns. Proteins with a highly similar sequence correlate well with the cellular localization site while those with dissimilar sequences indicate that they are distant and may or may not be colocalized. In 2002, Nair and Rost [24] [24] showed the correlation between sequence similarity with subcellular localization. They considered 11 different compartments and observed sequence conservation among the major compartments. BLAST, PSI-BLAST, and hidden Markov models (HMM) are routinely used for searching homologous sequences. The limitation of homology-based methods is more pronounced in cases where no homology is found between the query sequence and the annotated proteins sequence. Additionally, it is known that a single amino acid substitution in localization signals can change the localization of a protein [25][26][27][25,26,27]. Thus, sequence homology is a noncausal feature for the localization prediction and should be used with caution when applied to nonnative sequences or in case when homology is less [28].
3.3. Functional Motifs, Domains, and other Signatures Based Method
Proteins have evolved in different compartments, which limit their interactions with other proteins and ultimately impact their functions. Some of these proteins preserved some sequential or structural patterns or motifs. Though not all of these motifs and domains are specific to subcellular localization, many preferentially occur in some specific compartments and such domains can be used to predict the localization of any proteins. Studying proteins at a domain/motif level allows more accurate functional inference [29]. In 2002, Mott et al. [30] [30] first used 300 Simple Modular Architecture Research Tool (SMART) domains to predict three subcellular locations viz secreted, cytoplasm, and nucleus. After that, several works have been used for the protein motif and domains as features for protein localization predictions [31][32][31,32].
These motifs are not just limited to sequence patterns, but also extended to the structural information. There are a couple of tools such as PROSITE [33] [33] and MEME [34] that employed this feature to use for protein localization. While the structure is not available for a big chunk of protein sequences, this gap is filled by several proteins structure predictions servers, like I-TASSER and C-I-TASSER servers [35].[35]
3.4. Signal Peptide Based Method
Signal peptides are short amino acid sequences in the amino terminus of the newly synthesized proteins and are found in all organisms including bacteria, archaea, and eukaryotes. The function of the signal peptide is to enable the transport machinery to translocate the proteins to different subcellular locations. They are present in secretory proteins and in transmembrane proteins and the protein residing in different eukaryotic organelles have different types of signal peptide sequences [36]. The signal peptide is followed by a stretch of amino acids that form the cleavage site recognized by peptidases and the signal peptide is removed after translocation, except in the case of transmembrane proteins. In case of transmembrane proteins, this signal peptide serves as signal anchor sequences. The importance of various signal peptide sequences in proteins in their subcellular localization has led to attempts to predict the subcellular location on the basis of the signal peptide present in proteins. The prediction of the signal peptide involves two main tasks: (1) discriminating between the signal peptide and signal anchor sequences and (2) also predicting the position of the signal peptide cleavage site [37]. The major challenge in signal peptide prediction is discriminating between true signal sequences and other hydrophobic regions. In addition to it, the accurate prediction of the cleavage site is also very important due to the high variability of the signal sequence length and the absence of sequence motifs that unambiguously mark the position of the cutting site [37][38]. A number of prediction methods are available that recognize and predict the subcellular location on the basis of signal peptides (Table 1). SignalP was the first publicly available method [15] [15] and there are many versions available, which were developed based on different methods. Version-1 [39] was based on artificial neural networks, while version-2 [40] was based on hidden Markow models, version-3 [41] [41] has an improved cleavage site prediction, version-4 [42] has improved discrimination of signal peptides and TM helices, and version-5 [38] [38] is a deep neural network-based method combined with a conditional random field classification and an optimized transfer learning for improved signal peptide prediction.
Table 1.
Some useful signal peptide-based methods.
| Method | Tools Used | Performance Matrix | Locations/Organism | Availability | Year |
|---|---|---|---|---|---|
| SignalP-5.0 * | convolutional and recurrent (LSTM) neural networks | MCC, precision and recall | Archaea, Gram-positive Bacteria, Gram-negative Bacteria and Eukarya | http://www.cbs.dtu.dk/services/SignalP/ | 2019 |
| TargetP 2.0 * | recurrent neural networks (RNNs) network | Precision, recall, F1-score, MCC | mitochondrial, chloroplastic, secretory pathway | http://www.cbs.dtu.dk/services/TargetP/ | 2019 |
| SigUNet | Convolutional neural network | MCC, precision, recall, F1 measure | Eukaryotes, Gram-positive and Gram-negative bacteria | https://github.com/mbilab/SigUNet | 2019 |
| DeepSig | Convolutional Neural Networks | MCC, False Positive Rate, precision and recall | Eukaryotes, Gram-positive bacteria and Gram-negative bacteria | https://deepsig.biocomp.unibo.it | 2018 |
| SChloro | SVM | Accuracy, Recall, Precision, F1-score, and MCC | six chloroplastic sub-compartments | http://schloro.biocomp.unibo.it | 2017 |
| PredSL | combination of neural networks, Markov chains, scoring matrices (PrediSi), and HMMs, | Accuracy | Eukaryotic subcellular location | http://bioinformatics.biol.uoa.gr/PredSL/ | 2006 |
| TatP | HMM/artificial neural networks. | S-score and the C-score, Y-score, D-score | bacteria | http://www.cbs.dtu.dk/services/TatP/ | 2005 |
| ChloroP | Neural network | MCC, sensitivity, specificity | chloroplast transit peptides | http://www.cbs.dtu.dk/services/ChloroP/ | 1999 |
* There are different versions of the software available, but here we mentioned only the recent one.
The signal peptide-based method is a good approach to predict the proteins that contain the signal peptide, but it has some drawbacks, which make these methods not able to be applied for proteome scale prediction. (i) Not all proteins contain signal peptides. There are many proteins that do not have any reported signal peptide sequence and despite this are still translocated to their respective subcellular location. (ii) Many proteins follow the “piggyback import” mechanism during protein translocation, which means these proteins do not have any specific signal peptide for the localization, but they interact and bind to different proteins that have a signal peptide for translocation and then are co-imported to specific target locations [43][44][43,44].
3.5. Non-Sequence Derived Features
A variety of non-sequence derived features have been used to predict subcellular localization. For example, LOC3D [45], which used the structural information for identification and prediction of proteins subcellular locations. There are a number of non-sequence derived features that have been used in an automated classifiers including immunohistochemistry [3][46][47][3,46,47], fluorescence microscopy image [48][49][48,49], protein–protein interaction (PPI) data [50], expression data [51], and recommendation systems [52].
3.6. Integrated Method
The different strategies for predicting protein localization have their own strengths and weaknesses. To enhance the performance of prediction methods, it is important to combine multi-characteristic strategies, which give more complete information to understand the relationship between protein localization with its sequence, structure, physicochemical properties, and function. Hence a combination of different input vectors and different tools will be the successful strategy in protein subcellular localization prediction. Many methods have successfully utilized the combination of protein features to enhance the performance of protein subcellular localization predictions. The Protein Subcellular Localization Prediction Tool (PSORT) family method is one of the integrated methods, which contains several tools for localization prediction. The family includes a number of tools: (i) PSORT [53], the first integrated method of the PSORT family (http://psort.org) for the plant and bacterial protein, (ii) PSORT II [53] [54] for yeast and animal proteins, (iii) iPSORT [55] [55] for N-terminal sorting signals for plant or non-plants; (iv) PSORTb [56][57][58] [56,57,58] for bacterial and archaeal proteins, and (v) WoLF PSORT [59] [59] for eukaryotic proteins including plants, animals, and fungi.
A similar approach was taken by many researchers where they integrated biological or empirical sequence features correlated with subcellular location with a variety of machine-learning algorithm such as KNN, SVM, and deep learning: MultiLoc, integration of the phylogenetic profile and GO terms of retrieved homologues such as MultiLoc2, CELLO2.5, SherLoc2, YLoc, iLoc-Euk, Loctree3, DeepLoc, etc. People are also integrating different computational tools for predicting subcellular localization. The Bologna Unified Subcellular Component Annotator (BUSCA) [60] [60] is an example of such an integrated tool where the author combines methods for identifying signal and transit peptides (DeepSig and TPpred3), GPI-anchoes (PredGPI), and transmembrane domains (ENSEMBLE3.0 and BetAware) with tools for discriminating subcellular localization of both globular and membrane proteins (BaCelLo, MemLoci, and SChloro). This integrated method performs better than the other methods based on single feature approaches. There are a number of recently developed subcellular localization methods available, which are used by a wide range of researchers (Table 2 and Table S1).
Table 2. List of subcellular localization methods.
| Method | Tools Used | Performance Matrix | Feature Based | Locations/Organism | Availability | Year | ||
|---|---|---|---|---|---|---|---|---|
| DeepPred-SubMito | Convolutional neural network | Accuracy, MCC | Sequence information | Mitochondrial and submitochondrial proteins | https://github.com/jinyinping/DeepPred-SubMito.git | 2020 | ||
| SubMito-XGBoost | Extreme gradient boosting (XGBoost) | Sensitivity, Specificity, False positive rate, MCC, F1-measure, precision | Sequence information | Submitochondrial proteins | https://github.com/QUST-AIBBDRC/SubMito-XGBoost/ | 2020 | ||
| mRNALoc | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence information | eukaryotic | http://proteininformatics.org/mkumar/mrnaloc | 2020 | ||
| SCLpred-EMS | Convolutional neural network | Sensitivity, Specificity, False positive rate, MCC | Sequence information | endomembrane system and secretory pathway | http://distilldeep.ucd.ie/SCLpred2/ | 2020 | ||
| BUSCA | Integrated method of DeepSig, TPpred3, PredGPI, BetAware and ENSEMBLE3.0 | Precision, recall, F1-score, MCC | Sequence information, signal and transit peptides, glycophosphatidylinositol (GPI) anchors and transmembrane domains | Gram-positive, gram-negative, fungi, plant, animal | http://busca.biocomp.unibo.it | 2018 | ||
| SubMitoPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence and domain information | Mitochondrial and submitochondrial proteins | http://proteininformatics.org/mkumar/submitopred/ | 2018 | ||
| pLoc-mEuk | ML-GKR (multi-label Gaussian kernel regression) classifier | Coverage, Accuracy, Absolute true, Absolute false | Gene Ontology and Chou’s general PseAAC | 22 different subcellular localizations of eukaryotic proteins | http://www.jci-bioinfo.cn/pLoc-mEuk/ | 2018 | ||
| ERPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence information, | ER Proteins | http://proteininformatics.org/mkumar/erpred/index.html | 2017 | ||
| DeepLoc | deep recurrent neural networks | Accuracy, MCC | Sequence information | 10 different location of eukaryotic proteins | http://www.cbs.dtu.dk/services/DeepLoc | 2017 | ||
| SubNucPred | SVM | Sensitivity, Specificity, Accuracy, MCC | Sequence and domain information | Nuclear and subnuclear protein | http://proteininformatics.org/mkumar/subnucpred/ | 2014 | ||
| LocTree3 | SVM and homology | Accuracy, recall, standard deviation, standard error | Homology-based, Gene Ontology | 18 classes for eukaryotes, in six for bacteria and in three for archaea | http://www.rostlab.org/services/loctree3 | 2014 | ||
| PlantLoc | localization motif search | accuracy | localization motif information | 11 different location of plant proteins | http://cal.tongji.edu.cn/PlantLoc/ | 2013 | ||
| iLoc-Cell, package of predictors for subcellular locations of proteins. It includes iLoc-Hum, iLoc-Animal, iLoc-Plant, iLoc-Euk, iLoc-Virus, iLoc-Gpos, iLoc-Gneg |
multi-label learning, multi-label KNN | Accuracy, Precision, Recall, Absolute-true rate, Absolute-false rate, | Sequence information, gene ontology, PSSM, | Different subcellular location of Human, animals, plants, eukaryotic, Virus, gram-positive, gram-negatives | http://www.jci-bioinfo.cn/iLoc-Cell | 2011, 2012, 2013 | ||
| MARSpred | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information, PSSM | cytosolic and mitochondrial aminoacyl tRNA synthetase | http://www.imtech.res.in/raghava/marspred/ | 2012 | ||
| SCLPred | Neural Network | Sensitivity, specificity, False positive rate, MCC | primary sequence and multiple sequence alignments | four classes for animals and fungi and five classes for plants | http://distill.ucd.ie/distill/ | 2011 | ||
| AtSubP | SVM | Sensitivity, specificity, error rate, MCC, ROC curve | Sequence information, PSSM | subcellular localization of Arabidopsis | http://bioinfo3.noble.org/AtSubP | 2010 | ||
| Euk-mPLoc 2.0 | OET-KNN (Optimized Evidence-Theoretic K-Nearest Neighbor) classifiers | accuracy | gene ontology information, functional domain information, and sequential evolutionary information | eukaryotic proteins among the following 22 locations | http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/ | 2010 | ||
| PSORTb | SVM | Precision, recall, accuracy, MCC | Sequence information | Different subcellular location of Gram-negative, Gram-positive, archaea | http://www.psort.org/psortb | 2010 | ||
| YLoc | naïve Bayes alongside entropy-based discretization | overall accuracy, F1-score | Sequences information, GO-term and motif | animal, fungal and plant proteins | www.multiloc.org/YLoc | 2010 | ||
| SubChlo | evidence-theoretic | K | -nearest neighbor (ET-KNN) algorithm | overall accuracy, accuracy | Sequences information (PseAAC), | chloroplast proteins | http://bioinfo.au.tsinghua.edu.cn/subchlo | 2009 |
| MultiLoc2 | SVM | Sensitivity, specificity, Accuracy, MCC | phylogenetic profiles and gene ontology terms | Plant, Animal, Fungal | https://abi-services.informatik.uni-tuebingen.de/multiloc2/webloc.cgi | 2009 | ||
| AAIndexLoc | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information and physicochemical properties | Animal, Fungal and plants | http://aaindexloc.bii.a-star.edu.sg | 2008 | ||
| Cell-PLoc package of predictors for subcellular locations of proteins. It includes Euk-mPLoc, Hum-mPLoc, Plant-PLoc, Gpos-PLoc, Gneg-PLoc, Virus-PLoc | KNN or OET-KN algorithm | Accuracy and F1 score | GO and functional domain information | 22 subcellular location of eukaryotic, human, plant, Gram-positive bacterial, Gram-negative bacterial and viral proteins | http://chou.med.harvard.edu/bioinf/Cell-PLoc | 2008 | ||
| ProLoc-GO | SVM-GO, k-NN-GO and fuzzy k-NN-GO | MCC | GO term information | eukaryotic, human, | http://iclab.life.nctu.edu.tw/prolocgo | 2008 | ||
| ProLoc | SVM | Accuracy | physicochemical composition | subnuclear localizations | http://iclab.life.nctu.edu.tw/proloc | 2007 | ||
| SherLoc | SVM | Sensitivity, specificity, MCC | Sequence information | eukaryotic proteins | http://www-bs.informatik.uni-tuebingen.de/Services/SherLoc/ | 2007 | ||
| MitPred | SVM | Sensitivity, specificity, Accuracy, MCC | Sequence information | Mitochondrial proteins | http://www.imtech.res.in/raghava/mitpred/ | 2006 | ||
| BaCelLo | SVM | Coverage, Normalized Accuracy, geometric average , overall accuracy, Generalized Correlation |
Sequence information | Plant, Animal, Fungal | http://www.biocomp.unibo.it/bacell/ | 2006 | ||
| HSLpred | SVM | Accuracy, MCC, Reliability index | Sequence information | Human Protein | http://www.imtech.res.in/raghava/hslpred/ | 2005 | ||
| PSLpred | SVM | Accuracy, MCC, Reliability index | Sequence information | gram-negative bacterial proteins | http://www.imtech.res.in/raghava/pslpred/ | 2005 | ||
| ESLpred | SVM | Accuracy, MCC, Reliability index | Sequence information and PSSM | eukaryotic proteins | http://www.imtech.res.in/raghava/eslpred/ | 2004 |