Intracellular calcium (Ca2+) is an important second messenger that regulates multiple cellular functions, such as synaptic plasticity, action potentials, and learning and memory. Ca2+ dyshomeostasis, on the other hand, contributes to detrimental mechanisms such as necrosis, apoptosis, autophagy deficits, and neurodegeneration. Perturbations in intracellular Ca2+ are involved in many neurodegenerative diseases including Alzheimer's disease (AD), Parkinson’s disease, and Huntington’s disease [7]. Ca2+ dyshomeostasis is an early event in the AD timeline. Ca2+ dysregulation in AD comes as a result of hyperactivity of Ca2+ channels in the plasma membrane and intracellular compartments. It does not seem to be restricted to neurons, but rather is a global phenomenon that affects many cell types in the brain.
- calcium homeostasis
- Alzheimer’s disease
- endoplasmic reticulum
- mitochondria
- astrocytes
- microglia
1. Calcium Dysregulation Is a Hallmark of Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia, affecting more than 30 million people worldwide. It is characterized by accumulation of extracellular amyloid β (Aβ) plaques—or senile plaques—composed of Aβ peptide, intraneuronal fibrillary tangles (NFTs) comprising hyperphosphorylated and misfolded microtubule-associated protein tau, and selective neuronal loss, particularly in brain regions like the neocortex and hippocampus, eventually leading to memory loss and a decline in cognitive function. Most AD cases are sporadic (SAD), with less than 1% due to genetic mutations. Risk factors, such as aging, lifestyle, obesity, or diabetes, or genetic factors such as carrying the allele ε4 in the apolipoprotein E (ApoE) gene predispose individuals to SAD development [1]. Genetically inherited forms of AD (familial AD, FAD) show early onset and are caused by mutations in genes coding for presenilin (PS) 1, PS2, or amyloid precursor protein (APP), all involved in the Aβ generation pathway. Other than the onset, there are no clear differences regarding symptoms or histopathological features between SAD and FAD. Different hypotheses have been proposed to explain the origin of AD. The relation to genetics in FAD supported the “amyloid cascade hypothesis”, which suggests that AD pathogenesis is initiated by overproduction of Aβ and/or failure of its clearance mechanisms, upstream of tau dysregulation [2]. However, other hypotheses that explain the etiology of AD are being considered. The “cholinergic hypothesis” [3], “tau propagation hypothesis” [4], “inflammatory hypothesis” [5], or “glymphatic system hypothesis” [6] are among the most relevant.
Back in the mid-1980s, Khachaturian proposed that Ca2+ dysregulation led to neurodegeneration, suggesting that a sustained imbalance of cellular Ca2+ could disrupt normal neuronal functions and lead to neurodegenerative diseases such as AD [8][7][8]. Since then, many reports have shown Ca2+ dysregulation in AD (both in SAD [9,10][9][10] and in FAD [11]), animal models of the disorder [12[12][13][14][15][16][17][18][19],13,14,15,16,17,18,19], and cells from human AD patients [20]. The “Ca2+ hypothesis of Alzheimer’s disease” [21] postulates that activation of the amyloidogenic pathway causes a remodeling of normal neuronal Ca2+ signaling pathways, which then alters Ca2+ homeostasis and leads to the disruption of the mechanisms involved in learning and memory. Neuronal Ca2+ dyshomeostasis seems to manifest early in AD progression prior to the development of histopathological markers or clinical symptoms [22]. Similarly, AD is also marked by Ca2+ disruption in other cells in the brain such as astrocytes and microglia. Whether disruption of Ca2+ homeostasis is cause or consequence of AD pathology is still a matter of debate.
Up to date, there are only two types of Food and Drug Administration (FDA)-approved therapies for AD treatment (www.alzforum.org)—acetylcholinesterase inhibitors and N-methyl-d-aspartate receptor (NMDAR) antagonists—and neither can cure or reverse the disease, but can, at least, transiently relieve patients’ symptoms [23]. Unfortunately, drugs targeting Aβ have been mostly unsuccessful. Although these therapies have shown some success in clearing Aβ plaques from the AD brain, they have failed to relieve the cognitive decline of AD patients in clinical trials [24], with the exception of aducanumab, which demonstrated both clearance of plaques and modest gains in cognitive function [25]. In addition, the well-known lack of correlation between cognitive symptoms and Aβ deposition further supports the idea of the need for different approaches [26]. Ca2+ dyshomeostasis is an early molecular defect in AD and might precede Aβ and tau deposition [22].
2. Neuronal Ca2+ as a Therapeutic Target in AD
Ca2+ is a fundamental regulator of neuronal fate; thus, intracellular Ca2+ homeostasis must be finely tuned in physiological conditions. In the extracellular space, Ca2+ concentration is maintained between 1.1 and 1.4 mM, whereas resting cytosolic levels within neurons are maintained in the nM range (50–300 nM) [27]. After cell activation, intracellular Ca2+ concentrations increase rapidly to the μM range. This Ca2+ gradient allows the initiation of different signaling cascades. Ca2+ levels in the endoplasmic reticulum (ER) are nearly a thousand times higher than those of the cytoplasm [28]. Ca2+ signals are generated by the influx of Ca2+ from the extracellular space or by Ca2+ release from intracellular stores. Ca2+ enters neurons mainly through plasma membrane channels and is then buffered by Ca2+-binding proteins and organelles such as mitochondria. Even though the mechanisms responsible for neuronal Ca2+ dysregulation in AD are not completely understood, as discussed in the sections below, evidence shows that different compartments and/or organelles are involved (Figure 1).
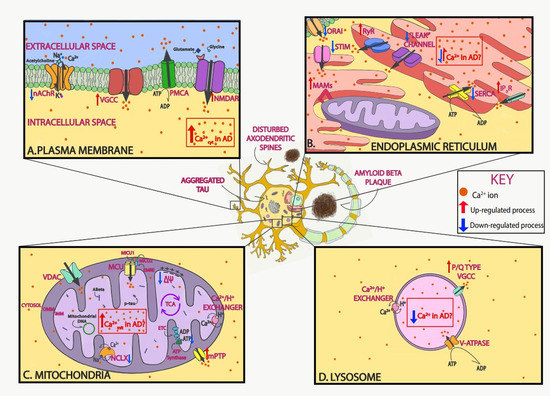
Figure 1. Neuronal Ca2+ as a therapeutic target in Alzheimer’s disease (AD). Schematic of Ca2+ dysregulation in neurons in AD that could be used as potential targets. In AD, Ca2+ dysregulation is present in many of the different compartments within neurons. In the plasma membrane, voltage-gated Ca2+ channels (VGCCs) and receptor operated Ca2+ channels, including N-methyl-d-aspartate receptors (NMDARs) and nicotinic acetylcholine receptors (nAChRs), allow for the influx of Ca2+ ions into the neuron after depolarization or ligand binding, respectively. Both Aβ and tau overactivate these channels and increase their function (A). In the endoplasmic reticulum (ER), Ca2+ is released via ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate receptors (IP3Rs) to the cytosol after stimulation. Ca2+ is then extruded by the sarco-endoplasmic reticulum ATPase (SERCA) pump, which actively consumes ATP while bringing Ca2+ into the lumen. AD-associated presenilin (PS) mutations impair IP3R and RyR signaling, increasing Ca2+ release into the cytosol, and diminish SERCA activity, increasing cytosolic Ca2+ concentration. Following ER Ca2+ depletion, the stromal-interacting molecule (STIM) interacts with the Orai channel in the plasma membrane to activate the store-operated Ca2+ entry (SOCE) pathway. SOCE is decreased by diverse familial AD (FAD) PS mutations and by soluble Aβ. Lastly, in order to facilitate the communication between mitochondria and ER, contact points known as mitochondrial-associated membranes (MAMs) are established. Increased association between the ER and mitochondria and enhanced Ca2+ transfer have been observed in AD (B). In the mitochondria, the voltage-dependent anion-selective channel protein (VDAC) lets Ca2+ across the outer mitochondrial membrane (OMM), and the mitochondrial Ca2+ uniporter (MCU) complex allows the influx of Ca2+ across the inner mitochondrial membrane (IMM). Ca2+ efflux is partially managed by the Na+/Ca2+ exchanger (NCLX). Both Aβ and tau (phospho-tau, p-tau) have been found in mitochondria. Elevated mitochondrial Ca2+ levels and decreased NCLX activity have been observed in AD (C). In the lysosome, the P/Q type VGCCs in their membrane regulate Ca2+ efflux into the cytosol, while the V-ATPase and Ca2+/H+ exchanger are in charge of lysosomal Ca2+ refilling (D). Additionally, Aβ and tau accumulate extracellularly and intracellularly, respectively, and lead to loss of dendritic spine density and synaptic function.
2.1. Targeting Plasma Membrane Receptors and Cytosolic Ca2+
Proper intracellular Ca2+ homeostasis is crucial for many neuronal functions. Disruption of this homeostasis might be one of the main mechanisms via which Aβ and tau exert their neurotoxicity. The main plasma membrane channels involved in neuronal Ca2+ influx from the extracellular space are voltage-gated Ca2+ channels (VGCCs), which allow Ca2+ influx following neuronal depolarization, and receptor-operated Ca2+ channels (ROCs), which open upon specific binding of the agonist, with NMDARs among the most important examples.
Toxic Aβ increases cytosolic Ca2+, which may affect a variety of enzymes (such as proteases or phosphatases), promote cytoskeletal modifications, cause the generation of free radicals, or trigger neuronal apoptosis [29]. It has been proposed that Aβ can overactivate channels and/or form pores in the cytosolic plasma membrane, allowing massive influx of Ca2+ from the extracellular space and increasing the overall Ca2+ levels in the cytosol, severely limiting normal cellular function [30,31][30][31]. Aβ potentiates Ca2+ influx through VGCCs, particularly L-type VGCCs. Excessive Ca2+ influx through these channels has been observed in cultured neurons following Aβ exposure and was shown to be blocked by the L-type VGCC inhibitor nimodipine [32]. This phenomenon, however, was not observed in brain slices from AD mouse models [33]. In addition, AD patients taking L-type VGCCs inhibitors such as nilvadipine (NILVAD multicenter trial) showed reduced Aβ levels but no improvement in cognitive decline [34,35][34][35]. Acute application of tau aggregates has also been observed to increase cytosolic Ca2+ and elevate reactive oxygen species (ROS) production via nicotinamide adenine dinucleotide phosphate (NADPH), an effect that can be prevented by nifedipine and verapamil, both L-type VGCC inhibitors [36]. This suggests that tau fibrils could also incorporate into the cell membrane to activate VGCCs and lead to neuronal dysfunction [36].
CALHM1 (Ca2+ homeostasis modulator protein 1) is a Ca2+ channel highly expressed in neurons in the hippocampus that allows cytosolic Ca2+ influx in response to decreases in extracellular Ca2+ [37]. Its activation triggers different kinase signaling cascades in neurons. The CALMH1 polymorphism P86L has been proposed as a risk factor for late-onset SAD [9,37][9][37], an argument that has been challenged by other groups [38,39][38][39]. Nevertheless, increased levels of Aβ have been observed in transfected cells expressing the P86L polymorphism, suggesting a role for CALMH1 in AD [37]. Additionally, the P86L polymorphism alters the channel permeability to Ca2+ [37]. A recent study demonstrated that CALHM1 deficiency in mice leads to cognitive and neuronal deficits, which manifest memory impairment and hippocampal long-term potentiation (LTP) [40] [40], pointing to CALHM1 as a potential treatment target in AD.
NMDARs are a subfamily of ionotropic glutamate receptors involved in the excitatory synaptic transmission and synaptic plasticity of the brain. Specific types of NMDARs are much more permeable to Ca2+ than other ionotropic glutamate receptors and are often implicated in neuronal pathophysiology. NMDARs are mainly composed of GluN2A and GluN2B in the brain areas most affected in AD [41]. Extra-synaptic GluN2B-containing NMDARs have been associated with excitotoxicity (the excessive neuronal death induced by cellular Ca2+ overload due to excessive stimulation of glutamate receptors) and the toxic effect of Aβ oligomers in AD [42,43][42][43]. For this reason, selective GluN2B subunit antagonists may be a strategy to prevent synaptic dysfunction in AD. Aβ42 peptides interact with NMDARs, potentiating their activity and leading to increased Ca2+ influx, thus contributing to the synapse loss observed in AD [44,45][44][45]. Additionally, as demonstrated in mouse models of AD, glutamate-induced excitotoxicity is inhibited by tau reduction [46] [46] and exacerbated by tau overexpression [47,48][47][48]. In turn, glutamate-induced excitotoxicity increases tau expression [49] [49] and phosphorylation [50], while activation of extra-synaptic NMDAR leads to tau overexpression, neuronal degeneration, and cell loss [51]. Memantine—a weak NMDAR antagonist—is one of the two FDA-approved drugs to treat AD patients and the only NMDAR antagonist [23]. It provides modest improvements to memory and cognitive performance in moderate to severe AD patients [52,53][52][53]. Memantine restricts excessive Ca2+ influx, thus reducing neuronal excitotoxicity, and, due to its low activity, the basal NMDAR function is preserved. Memantine has also shown neuroprotective effects against oxidative stress, neuroinflammation, and tau phosphorylation [54,55][54][55].
Ionotropic neuronal nicotinic acetylcholine receptors (nAChRs) respond to the neurotransmitter acetylcholine (ACh) and to drugs such as the agonist nicotine. They are permeable to Na+, K+, and Ca2+. The nAChRs expressing the α7 subunit have the highest conductance for Ca2+ and are found in brain regions most susceptible to AD [56]. In the basal forebrain, cholinergic neuronal loss and decreased levels of ACh mediate cholinergic impairment, which eventually leads to short-term memory loss [57,58,59][57][58][59]. The loss of cholinergic innervation in early AD led to the “cholinergic hypothesis of AD” [3]. Galantamine and rivastigmine (for use in mild to moderate AD) and donepezil (in mild to severe AD) are the cholinesterase inhibitors FDA-approved to treat AD [23]. These drugs act by increasing ACh levels, which delay the progression of AD through Ca2+-dependent mechanisms. Furthermore, supplemented with memantine, it has been proposed that this combination could provide greater benefits on behavior, cognition, and global outcomes in AD [60].
Exposure of hippocampal and cortical neurons to tau also increased intracellular Ca2+ levels through muscarinic receptors [61]. Interestingly, Ca2+ activates many kinases, including those responsible for tau phosphorylation—such as glycogen synthase kinase 3β (GSK3β)—and, therefore, Ca2+ dyshomeostasis may increase tau phosphorylation and NFT formation [62] [62]. Given that tau pathology correlates better with cognitive impairments than Aβ deposition, tau targeting is expected to be more effective once clinical symptoms emerge [63]. It has long been known that tau-expressing cells secrete normal and pathological tau [64], which can be taken up by other cells, seeding and spreading tau pathology [4,65,66,67][4][65][66][67]. Led by immunotherapy approaches, the efforts to target tau with therapeutics focus on reducing tau pathology by limiting the spread of extracellular tau across brain regions [68]. Anti-tau immunotherapy has shown potential in numerous clinical studies. Both active and passive tau immunization seem to offer a promising option by reducing tau pathology [69]. Active tau immunization, however, seems to elicit a risk of adverse immune reactions from targeting the normal protein. Other tested approaches involve reducing tau expression (with small interfering RNA or antisense oligonucleotides; siRNA and ASOs, respectively), targeting tau modifications, reducing tau aggregation, and stabilizing microtubules. Preventing or reducing pathologic tau has been shown to improve cognitive and motor impairments in animal models with neurofibrillary pathology, and several tau antibodies and vaccines have been tested in preclinical studies in the last years. Immunotherapy is currently at the stage of drug development (recently reviewed in [68,69,70][68][69][70]), and, as of today, eight humanized tau antibodies and two tau vaccines are under clinical trial for AD or frontotemporal dementia [71] [71] (www.alzforum.org).
The use of intravital imaging and transgenic mouse models of AD have allowed for direct observation of cytosolic Ca2+ dysregulation. In vivo, neuronal cytosolic Ca2+ dyshomeostasis is more likely to be observed in the vicinity of amyloid β plaques, but is detectable in neurons throughout the cortex [15]. Higher Ca2+ levels were observed in neurons close to amyloid plaques in a commonly used mouse model of cerebral amyloidosis (APPSwexPS1ΔE9, APP/PS1) [15], but only after plaque deposition and not before. Cytosolic Ca2+ overload was absent in mice harboring only the PS1 mutation (typically lacking plaque deposition). The mechanisms of Ca2+ dysregulation involved activation of calcineurin (CaN), a Ca2+/calmodulin-dependent protein phosphatase sensitive to subtle rises in intracellular Ca2+ levels, and whose activation induces long-term depression (LTD). Ca2+ dysregulation in neurites was linked to neurodegeneration (neuritic blebbing and beading), which can be partially prevented by inhibiting CaN [15]. Elevated Ca2+ levels in the neurites impair synapses, by increasing the frequency of spontaneous synaptic potentials and reducing plasticity. In addition, pathological increases in neuronal network activity—observed as increased frequencies of somatic Ca2+ transients—potentiate Aβ release into the extracellular space [72,73][72][73]. In the APP23/PS45 mouse model of AD (overexpressing mutant APPSwe and mutant PS1G348A), neuronal hyperactivity was observed around amyloid plaques in the cortex, only after plaque deposition [14]. Hyperactive neurons, however, were found in the CA1 region of the hippocampus in pre-depositing animals [13]. Direct application of soluble Aβ onto the wild-type (Wt) naïve brain increased cytosolic Ca2+ levels [12] [12] and induced neuronal hyperactivity [13]. Acute treatment with the γ-secretase inhibitor LY-411575, which reduces soluble Aβ levels, normalized the frequency of Ca2+ transients prior to plaque deposition [13].
Interestingly, AD patients are more prone to developing epileptic seizures [74]. Blocking network hyperactivity with the antiepileptic drug levetiracetam improves learning and memory, reverses behavioral abnormalities, and reverts synaptic deficits in the hippocampus in an AD mouse model [75]. In the same way, it has been observed that tau is implicated in neuronal circuit deficits in mouse models of AD expressing both Aβ and tau. Tau effects dominate those of Aβ and are mostly dependent on the presence of soluble tau [16]. According to the authors, this dramatic effect could suggest a possible cellular explanation contributing to disappointing results of anti-Aβ therapeutic trials. This abnormal network activity and its resultant AD-related cognitive deficits in mice point to neuronal hyperactivity as a promising therapeutic target in AD.
Aducanumab is a high-affinity, fully human immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds to aggregated Aβ fibrils and soluble oligomers (and not monomers) in the brain parenchyma [25]. It was shown that it could ameliorate Ca2+ dysregulation in AD. Using multiphoton microscopy and a Ca2+ reporter, it was observed that a single topical application of the antibody onto the brain surface of mice depositing amyloid plaques (Tg2576 AD model) led to a reduction in existing amyloid deposits [76]. Peripheral administration of the antibody over a period of 6 months rescued Ca2+ overload in transgenic neurites, restoring them to control levels within 2 weeks. The authors suggested that aducanumab exerted its function by targeting amyloid deposits, including soluble oligomeric Aβ [76]. In March 2019, the termination of all aducanumab clinical trials was announced after an interim analysis of EMERGE and ENGAGE trials predicted the phase III placebo-controlled studies would not meet their primary end points. However, in a subsequent analysis of a larger dataset from the EMERGE trial, aducanumab met the primary end point, and the FDA accepted the aducanumab application for review [77]. If the case is approved, aducanumab would be the first drug to combat the root causes of AD.
2.2. Targeting ER Ca2+ and SOCE
The ER is an important subcellular organelle involved in protein synthesis, modification, and folding. Additionally, it is a dominant Ca2+ reservoir in the cell, critical for maintaining intracellular Ca2+ levels [27]. Ca2+ is released from the ER after activation of either inositol 1,4,5-trisphosphate receptors (IP3Rs) or ryanodine receptors (RyRs). Ca2+ efflux from the ER modulates a range of neuronal processes, including regulation of axodendritic growth and morphology or synaptic vesicle release [78]. The sarco-endoplasmic reticulum ATPase (SERCA) pump, which actively consumes ATP, is important for extruding Ca2+ into the ER lumen, where it is sequestered by binding to proteins such as calsequestrin and calretinin, priming this organelle as a critical component of Ca2+ buffering.
Impaired IP3R signaling in the ER was an early discovery in AD. It was shown that human cells from FAD patients exhibited enhanced Ca2+ release in response to IP3R-generating stimuli [79]. Fibroblasts from asymptomatic members of AD families [80], as well as PS1 knock-in mice and other presymptomatic AD mouse models, showed the same enhancement [81]. These observations suggested that FAD mutations contribute to Ca2+ dysregulation, even before pathology deposition or cognitive impairments were evident. A reduction in IP3R expression can normalize Ca2+ homeostasis and restore hippocampal LTP in mouse models of AD [82]. PSs are transmembrane proteins found in the ER membranes and form the catalytic core of the γ-secretase complex that processes APP and other type 1 transmembrane proteins, such as Notch [83,84][83][84]. PSs are essential for learning and memory, as well as neuronal survival during aging in the murine cerebral cortex [85,86][85][86]. Mutations in PSs have been shown to affect APP processing, leading to increased production of the more hydrophobic neurotoxic form Aβ42 [87][88][89] [87,88,89] or increasing the Aβ42/40 production ratio [90]. It has also been proposed that PSEN mutations cause a loss of presenilin function in the brain, triggering neurodegeneration and dementia in FAD [91]. Mutations in PS1 and PS2 might stimulate IP3Rs, leading to exaggerated Ca2+ release through these channels [79,80,81][79][80][81]. An alternative hypothesis suggested that PSs function as passive low-conductance leak channels in the ER membrane. AD-associated PS mutations might impair this leak function, resulting in ER Ca2+ overload [92] and leading to exaggerated increases in cytosolic Ca2+ upon stimulation of Ca2+ release. However, these observations have not been supported by other groups [93[93][94],94], and, despite extensive research, this subject is still a matter of controversy. Recently, it was proposed that the ER-based transmembrane and coiled-coil domain TMCO1 could be responsible for the ER Ca2+ leak [95].
RyR Ca2+ dysregulation was also observed before the histopathology and cognitive decline in AD. Both human brain tissue from AD patients and AD mouse models have shown increased expression of RyR (particularly RyR2) in affected brain regions in AD [96]. Exaggerated Ca2+ release from RyR has been related to impaired neurophysiology and synaptic signaling events, contributing to memory impairment in AD [33,97,98][97][98]. The FAD PS mutations also exaggerate Ca2+ release through the RyR, as a result of either increased expression of RyRs or sensitization of the channel activity [99,100][99][100]. Furthermore, RyR-mediated Ca2+ release upregulates secretases, increasing APP cleavage, Aβ fragments, and plaque deposition, and its blockage leads to Aβ reduction and improved memory impairment [101]. RyRs can also themselves be activated by Ca2+, which amplify IP3R activity via Ca2+-induced Ca2+ release mechanisms [102]. Additionally, Aβ aggregates themselves trigger ER Ca2+ release through IP3Rs and RyRs [103,104][103][104]. Recently, stabilization of RyR2 macromolecular complex by S107 (Rycal)—a benzothiazepine that prevents the dissociation calstabin2 from the RyR2 complex—showed therapeutic potential in vitro and in mouse models of AD in vivo. Application or administration of S107 reversed ER Ca2+ leak, reduced APP cleavage and Aβ production, and restored synaptic plasticity and cognitive deficits [105,106][105][106].
It has been found that, in cells lacking PS1, PS2, or PS1/2, or cells expressing either PS2 or FAD-PS2, SERCA activity is diminished, resulting in increased cytosolic Ca2+ [107]. Conversely, other studies have shown that mutations in PS influence SERCA by accelerating Ca2+ sequestration via ATPase [108], leading to an overfilled ER. In any case, these data suggest that normal PSs are required for normal SERCA functioning and suggest that PSs are a candidate target for development of therapeutics, independent of their role in APP processing.
Increased Ca2+ release from intracellular ER Ca2+ stores might exacerbate disease-mediated pathology. Accordingly, dantrolene, a negative allosteric modulator of RyR—and its central nervous system (CNS)-penetrant version Ryanodex—has been shown to reduce amyloid pathology, normalize ER Ca2+ homeostasis, restore synaptic structure and density, normalize synaptic plasticity, and improve behavioral performance in mouse models of AD [101,109,110][101][109][110]. This builds on RyR as a therapeutic target for AD, and further emphasizes the role of dysregulated ER Ca2+ as a key component in the AD pathogenesis.
ER Ca2+ depletion triggers a sustained extracellular Ca2+ influx to the cytosol through the store-operated Ca2+ entry (SOCE) pathway by activating STIM (stromal-interacting molecule) protein—which senses low Ca2+ concentration upon depletion of the ER stores—and plasma membrane channels Orai and TRPC (transient receptor potential canonical) [111]. Two forms of STIM are expressed in the brain (STIM1, predominantly in the cerebellum, and STIM2 in the hippocampus and cortex) [112]. SOCE refills the ER, keeping it ready for the next ER Ca2+ signal [113]. Disrupted SOCE has been observed in AD. SOCE is decreased by diverse FAD PS mutations [114,115][114][115] and in the presence of soluble Aβ [116]. It has also been proposed that SOCE deficits may be due to the decreased expression of STIM1 and/or STIM2 in FAD-linked PS1 mutations [117]. Related to this, overexpression of the dominant negative PS1 variant potentiates SOCE [118]. It has also been proposed that SOCE deficits might result from overfilled ER Ca2+ stores [114]. These findings, however, are inconsistent, as other groups have observed no differences or decreased ER Ca2+ concentration in mutant PS expressing cells [11,93,107][11][93][107]. Recently, it has been proposed that neuronal SOCE is required for maintaining the morphology of mushroom spines, modulating Aβ production and promoting memory functions [117,119][117][119]. Ca2+ entry via SOCE activates Ca2+/CaM-dependent kinase II (CamKII), which is upstream of gene transcription for maintenance of mature spines. Attenuated SOCE-mediated Ca2+ influx might reduce CaMKII activity while inducing destabilization of mushroom spines. This can reduce LTP-mediated memory formation [117]. Attenuated SOCE may also lead to inadequate ER refill, which might induce neuronal cell death via apoptosis [120,121][120][121]. STIM2 overexpression in AD models restores spine morphology, implicating SOCE in AD [122] [122] and suggesting that targeting SOCE in AD may avoid or restore dendritic spine loss. Additionally, it was recently found that expression of TRPC1, a subfamily of TRPCs, is decreased in AD cells and mouse models. While deletion of TRPC1 did not impair cognitive function or lead to cell death in physiological conditions, it did exacerbate memory deficits and increase neuronal apoptosis induced by Aβ. On the contrary, overexpression of TRPC1 inhibited Aβ production and decreased apoptosis [123]. Together, these studies suggest another mechanistic target for therapeutic development within the Ca2+ hypothesis of AD.
2.3. Targeting Mitochondrial Ca2+
Mitochondria are crucial organelles that provide energy to the cell in the form of adenosine triphosphate (ATP) via the process of oxidative phosphorylation. Mitochondria form a dynamic tubular network that extends throughout the cytosol, undergoing fusion and fission, which regulates the morphology and structure of the mitochondrial network [124]. Neurons rely strictly on mitochondria to produce ATP, with mitochondria being recruited in areas like synapses, where high energy is required. Mitochondria also buffer Ca2+ and shape its signal [125], which is involved in neurotransmission and maintenance of the membrane potential along the axon. At the synaptic level, mitochondria regulate the Ca2+ levels necessary for synaptic functions [126]. Mitochondrial Ca2+ uptake activates some dehydrogenases at the electron transport chain (ETC), activating mitochondrial respiration and ATP production [127]. The electrochemical gradient created by the ETC allows mitochondria to take up Ca2+. This mitochondrial Ca2+ participates in signal transduction and the production of energy. Mitochondria contain two major membranes, the outer mitochondrial membrane (OMM), which contains voltage-dependent anion-selective channel protein (VDAC), permeable to most molecules, and the inner mitochondrial membrane (IMM), which is impermeable to molecules and ions, unless they contain specific channels or transporters.
Ca2+ is taken up into the mitochondrial matrix through the mitochondrial Ca2+ uniporter (MCU) complex, a highly Ca2+-sensitive ion conductance channel [128,129][128][129]. The MCU is a macromolecular complex of proteins, which includes the pore and several regulatory subunits. It is ubiquitously expressed among organisms and defines the pore domain of the complex [128]. Two other proteins participate in the Ca2+ permeant pore: MCUb, whose expression is restricted to most vertebrates [130[130][131],131], and the essential MCU regulator (EMRE) [132]. The response of the MCU to extramitochondrial Ca2+ is regulated by the mitochondrial Ca2+ uptake (MICU) family of proteins, which are in the intermembrane space. MICU1 and MICU2 act as Ca2+ sensors, each with two Ca2+-binding EF-hand motifs that confer sensitivity to Ca2+ [133]. MICU1 and MICU2 also act as gatekeepers of MCU [134], with MICU1 getting involved when the extramitochondrial Ca2+ concentration is high, activating the channel open state. At low concentrations, the main player seems to be MICU2, leading to minimal accumulation of Ca2+ within mitochondria [135[135][136],136], thus preventing mitochondrial Ca2+ overload at resting conditions. MICU3, a paralog of MICU1 and MICU2, is mainly expressed in the CNS [137], and has been proposed to enhance mitochondrial Ca2+ uptake in neurons [138]. In regulating the MCU pore, the mitochondrial Ca2+ uniporter regulator 1 (MCUR1) also plays a role [139]. It has been suggested as a necessary player in MCU-mediated mitochondrial Ca2+ uptake. The small Ca2+-binding mitochondrial carrier protein (SCaMC, also known as SLC25A23) [140] [140] seems to also participate in the mitochondrial Ca2+ uptake by interacting with MCU and M1CU1.
Mitochondrial Ca2+ efflux occurs via the Na+/Ca2+ exchanger (NCLX) [141] [141] and leucine zipper- and EF hand-containing transmembrane protein 1 (Letm1), located at the IMM [142]. Excessive Ca2+ in the mitochondrial matrix induces the activation of the mitochondrial permeability transition pore (mPTP) and allows the release of Ca2+ ions and small molecules such as cytochrome c [143]. Mitochondrial Ca2+ levels are tightly regulated since excessive levels of Ca2+ within mitochondria, i.e., mitochondrial Ca2+ overload, result in the impairment of mitochondrial function, suppression of ATP production, increase in reactive oxygen species (ROS) production, and mPTP opening. This can lead to caspase activation and cell death via apoptosis [144].
Mitochondrial function has long been considered one of the intracellular processes compromised at the early stages in AD and likely in other neurodegenerative diseases. Moreover, the “mitochondrial cascade hypothesis” was proposed to explain the onset of SAD [145], which posits that mitochondrial dysfunction is the primary process to trigger the cascade of events that lead to late-onset AD. Even though the validity of this hypothesis has yet to be demonstrated, numerous mitochondrial functions are disrupted in AD [146], including mitochondrial morphology and number [147], oxidative phosphorylation, mitochondrial membrane potential, ROS production [148], mitochondrial DNA (mtDNA) oxidation and mutation [149], mitochondrial–ER contacts [150], and mitochondrial dynamics, including mitochondrial transport along the axon and mitophagy [151]. Additionally, both Aβ and tau have been found in mitochondria. Aβ is imported to mitochondrial matrix via translocase of the outer membrane (TOM) [152], and a fraction of intracellular tau has been found within the inner mitochondrial space [153]. Once in mitochondria, they interact with specific intramitochondrial targets, leading to the dysfunction of the organelle. Furthermore, tau accumulation in mitochondrial synaptosomes has been proposed to correlate with synaptic loss in AD brains [154].
Mitochondrial Ca2+ dysregulation is considered a fingerprint of AD. Mitochondrial Ca2+ overload can be a result of three different processes: (i) increased mitochondrial Ca2+ influx (following Ca2+ influx from extracellular space or Ca2+ transfer from ER), (ii) decreased mitochondrial Ca2+ efflux through NCLX, or (iii) reduced mitochondrial Ca2+ buffering. Neurotoxic Aβ can lead to mitochondrial Ca2+ overload, as shown in in vitro and in vivo models [155,156,157] [155][156][157]. Primary neurons in culture exposed to Aβ oligomers triggered mitochondrial Ca2+ overload, leading to mPTP opening, release of cytochrome c, and cell death via mitochondrial-mediated apoptosis [156] [156]. Additionally, studies in mouse neuroblastoma N2a cells co-transfected with the Swedish mutant APP and Δ9 deleted PS1 showed similar mitochondrial impairment, evidenced by the increased mitochondrial apoptotic pathway and caspase-3 activity [158]. Furthermore, Aβ can interact with cyclophilin D—a regulator of mPTP—and promote the release of cytochrome c through the opening of mPTP [159]. This causes neuronal injury and decline of cognitive functions, as shown in a mouse model of AD. Genetic deletion of CypD in Tg AD mice rescues mitochondrial impairment and improves learning and memory [160], suggesting that CypD could represent a potential therapeutic target in AD.
Recently, we showed mitochondrial Ca2+ overload in a mouse model of cerebral amyloidosis (APP/PS1). Using in vivo multiphoton imaging and a ratiometric Ca2+ reporter, we demonstrated increased levels of mitochondrial Ca2+ following Aβ deposition, which preceded neuronal cell death. Moreover, naturally secreted soluble oligomers applied to the healthy brain of Wt mice also increased mitochondrial Ca2+ levels, a process that could be prevented by MCU inhibition with the specific channel blocker Ru360 [157]. We also showed, for the first time, that the expression of mitochondrial Ca2+ transport-related genes in brain tissue from AD patients was impaired compared to control cases. In particular, genes involved in mitochondrial Ca2+ uptake (MCU complex) were downregulated, whereas the only one encoding for Ca2+ efflux (NCLX) was upregulated, suggesting a compensatory response to prevent mitochondrial Ca2+ overload [157]. However, others reported that different techniques used for evaluating expression showed conflicting results [161]. Another mechanism proposed for mitochondrial Ca2+ overload in AD is impairment of mitochondrial Ca2+ efflux. Loss of NCLX expression and functionality has also been suggested in AD, whereas genetic rescue of NCLX expression in neurons restored cognitive decline and cellular impairment in transgenic mouse models of AD [161]. Additionally, the more general cytosolic Ca2+ overload observed in vivo (as previously cited) may contribute to the observed mitochondrial Ca2+ overload. These observations suggest that restoring mitochondrial Ca2+ levels in AD could be a promising new therapeutic target against AD.
It has been previously proposed that nonsteroidal anti-inflammatory drugs (NSAIDs) may help in preventing the cognitive decline associated with aging [162]. Unfortunately, results from several clinical trials have given rather pessimistic results [163[163][164][165],164,165], partly due to inadequate CNS drug penetration of existing NSAIDs, suboptimal doses, unknown molecular targets (and, therefore, unknown pharmacodynamics), and toxicities. Nevertheless, in vitro studies have shown that NSAIDs such as salicylate and the enantiomer (R)-Flurbiprofen lacking anti-inflammatory activity, at low concentrations, are able to depolarize mitochondria and inhibit the driving force for mitochondrial Ca2+ uptake [166,167][166][167]. They act as mild mitochondrial uncouplers without altering cytosolic Ca2+ levels. This mild mitochondrial depolarization was able to prevent NMDA- and Aβ-induced mitochondrial Ca2+ uptake and cell death [155,156,168][155][156][168]. These results point to mitochondrial Ca2+ as a key player in Aβ-driven neurotoxicity and suggest a new mechanism of neuroprotection by NSAIDs independent of their anti-inflammatory activity. Another compound, TG-2112x, has been recently suggested as neuroprotective and proposed as a new therapeutic opportunity. Tg-2112x partially inhibits mitochondrial Ca2+ uptake without affecting the mitochondrial membrane potential or mitochondrial bioenergetics, protecting neurons against glutamate excitotoxicity [169].
Abnormal tau hyperphosphorylation also influences mitochondrial transport along the neuronal axon, which leads to a reduction in and impairment of mitochondria at the presynaptic terminal with detrimental consequences and eventual cell death [170,171][170][171]. In vitro and in vivo studies have shown that tau dysregulates Ca2+ homeostasis in mitochondria. Mitochondrial Ca2+ buffering and homeostasis are disrupted in cells overexpressing tau and those exposed to extracellular tau aggregates [61,172][61][172]. Additionally, basal mitochondrial Ca2+ levels have been shown to be elevated in patient-derived human induced pluripotent stem cell (iPSC) neurons expressing a tau mutation, likely due to the inhibition of NCLX by tau [173]. Elevation in mitochondrial Ca2+ levels by tau also increased the vulnerability to Ca2+-induced cell death [173]. Phosphorylated tau has also been found to interact with VDAC in AD brains, leading to mitochondrial dysfunction [174].
Mitochondria and ER membranes are juxtaposed and establish contact points known as mitochondrial-associated membranes (MAMs). They are dynamic lipid rafts enriched in cholesterol and sphingomyelin, as well as in proteins associated with Ca2+ dynamics [175,176][175][176]. MAMs allow for communication between ER and mitochondria, including metabolic pathways and Ca2+ transfer from ER to mitochondria [177]. Increased contacts between ER and mitochondria have been found in human fibroblast cells derived from FAD patients, human brain tissue, and AD mouse models [178,179][178][179]. An increased association between the ER and mitochondria has also been observed in a Tg mouse model of tauopathy [180]. Increased contact promotes mitochondrial bioenergetics, but excessive Ca2+ transfer can contribute to mitochondrial Ca2+ overload and suppression of normal mitochondrial functions, and Aβ oligomers have been found to induce massive Ca2+ transfer from ER to mitochondria [116,181,182,183][116][181][182][183].
Mitochondria-targeted protective compounds that prevent or minimize mitochondrial dysfunction could represent potential therapeutic strategies in the prevention or treatment of AD. However, several compounds targeting mitochondrial function have been tested in AD without a favorable outcome [184]. Nevertheless, the idea of AD as a multifactorial disease is widespread, and mitochondria as a therapeutic target combined with other medications is emerging as a valid therapy for AD. The list of pharmacologic approaches that directly target mitochondria includes antioxidants (such as vitamin E and C, coenzyme Q10, mitoQ, and melatonin) and phenylpropanoids (such as resveratrol, quercetin, or curcumin) [185]. Antioxidants are generally used to decrease oxidative stress and slow the progression of symptoms that generally accompany AD. Antioxidants such as coenzyme Q10 and mitoquinone mesylate (MitoQ) are antioxidants that directly target mitochondria [186]. Currently, there is a small clinical trial testing MitoQ on cerebrovascular blood flow in AD [187]. The Szeto-Schiller (SS) tetrapeptides, an alternative type of antioxidants that target mitochondria, are small molecules that can reach the mitochondrial matrix and act as antioxidants [188]. Specifically, SS31 (also known as elamipretide) selectively binds to cardiolipin and promotes electron transport while optimizing mitochondrial ATP synthesis [189]. In addition, SS31 inhibits mitochondria swelling and oxidative cell death. In mouse models of cerebral amyloidosis, it was shown that SS31 reduces Aβ production and mitochondrial dysfunction, and enhances mitochondrial biogenesis and synaptic activity [190]. Recently, SS31 combined with the mitochondrial division inhibitor 1 (Mdivi1) was tested in vitro with a positive outcome, suggesting this combination as a possible type of mitochondria-targeted antioxidant in AD [191]. Ongoing clinical trials regarding mitochondria in AD are reviewed in [187,192][187][192] and at www.clinicaltrials.gov.
2.4. Targeting Lysosomal Ca2+
Lysosomes are acidic organelles that participate in the endolysosomal system. They are important for autophagy and intracellular Ca2+ storage (with comparable Ca2+ levels to those of the ER) [193]. The Ca2+ transport in and out of the lysosomal lumen provides signals that modulate the fusion of autophagosomes and lysosomes. In order to maintain lysosomal Ca2+ homeostasis, lysosomes contain P/Q type VGCCs expressed in the lysosomal membrane that provide Ca2+ to the cytosol. Dysregulation in lysosomal Ca2+ release via VGCCs leads to defective autophagic fusion and flux [194]. The vacuolar-type H+-ATPase (V-ATPase) and Ca2+/H+ exchanger are in charge of lysosomal Ca2+ refilling [195]. It has been suggested that this refilling is largely dependent on ER Ca2+ [196]. V-ATPase activity predominantly maintains lysosomal pH; however, other ion channels localized to the lysosomal membrane participate in pH regulation during lysosomal proteolysis, including the chloride channel CLC7 [197] and the Ca2+ channel TRPML1 (mucolipin) [198,199][198][199]. Additionally, Ca2+ microdomains generated at the mouth of these channels have been suggested to take part in the regulation of autophagy [200].
Lysosomal Ca2+ efflux has been linked to changes in lysosomal pH. Recent reports suggested that decreased lysosomal Ca2+ in AD-linked mutations or PS1 knockout (KO) cells is a consequence of elevated lysosomal pH [201]. Raising lysosomal pH leads to autophagy defects and lysosomal Ca2+ efflux. PS1 mutant cells exhibit these defects. PS1 KO cells show deficiencies in lysosomal V-ATPase content and function, defective autophagy, and abnormal Ca2+ efflux. [201] [201]. Reversal of lysosomal pH abnormalities in PS1 KO cells, but not Ca2+ efflux deficits, was sufficient to rescue these same deficits [201]. These data suggest that lysosomal Ca2+ defects are secondary to lysosomal pH elevation, and that lysosomal Ca2+ dyshomeostasis contributes significantly to the overall Ca2+ dysregulation observed in PS1-deficient cells. However, other studies do not support these arguments, citing that, although the autophagosome and lysosome accumulation was apparent in PS1 or PS2 cells, defective lysosome acidification was not found [202]. In addition, defects in lysosome acidification or Ca2+ homeostasis have not been observed in FAD-PS2 models [203]. On the contrary, other studies have shown both reduced cytosolic Ca2+ signal and lower ER content in FAD-PS2 models. In particular, it was proposed that FAD-PS2 decreases ER and cis–medial Golgi Ca2+ levels by reducing SERCA activity, which could lead to defective autophagosome–lysosome fusion [203] [203]. Further studies are necessary to confirm these observations and demonstrate whether or not lysosomal Ca2+ or pH could be potential therapeutic targets for AD.
Autophagy is a lysosomal degradative pathway responsible for the recycling of different cellular constituents. Especially important under conditions of metabolic stress, this pathway aids in the cellular turnover of damaged or obsolete organelles in order to eliminate misfolded and aggregated proteins left behind by the ubiquitin-proteasome system [204]. Materials are engulfed within double-membrane vesicles (autophagosomes) and targeted to lysosomes for degradation of molecular components. Disruption of autophagy results in accumulation of autophagic vacuoles within swollen dystrophic neurites of affected neurons [205]. Lysosomal Ca2+ has been proposed to trigger transcriptional activation of autophagic proteins [200]. Impairment of the autophagy–lysosomal pathway has been described as a hallmark of AD related to lysosomal Ca2+ dyshomeostasis. This dysregulation impacts clearance of Aβ and hyperphosphorylated tau, and contributes to their accumulation in the brain [206,207] [205][206].
3. Astrocytic Ca2+ as a Therapeutic Target in AD
Astrocytes, the most abundant cells in the brain, are key regulators of molecular homeostasis in the nervous system. They provide trophic and metabolic support to neurons, sense and modulate neuronal network excitability, and participate in neurovascular coupling and maintenance of the blood–brain barrier [208,209,210][207][208][209][210]. Astrocytes do not generate action potentials, but exhibit Ca2+ transients followed by a release of gliotrasmitters—such as ATP, glutamate, or gamma-aminobutyric acid (GABA)—in response to neurotransmitters [211]. It has been proposed that the Ca2+ global signals—propagating waves—rely on Ca2+ release from the ER (mostly mediated by IP3R). Local Ca2+ microdomains, on the other hand, result from Ca2+ influx via ionotropic receptors, TRPs, SOCE, mitochondrial Ca2+ activity, or reversed Na+/Ca2+ exchangers [212].
In AD, astrocytes become activated. Reactive astrogliosis is characterized by the biochemical, functional, and morphological reshaping of astrocytes aimed at neuroprotection [213]. Reactive astrocytes upregulate activation markers such as glial fibrillary acidic protein and vimentin. Using postmortem human tissue, it has been shown that reactive astrocytes associated with plaques express higher levels of the glutamate metabotropic receptor mGluR5, which induces Ca2+ release form intracellular stores [214]. In vitro, exposure of astrocytes to Aβ increases basal intracellular Ca2+ levels as a result of extracellular Ca2+ entry, release from mGluR5 and IP3R, and induced Ca2+ oscillations or transients [214,215][214][215]. Pharmacological inhibition of ER Ca2+ release blocks the Aβ-induced astrogliosis both in cultured astrocytes and in organotypic slices [216]. As observed in co-cultures of neurons and astrocytes, the Aβ-induced astrocytic Ca2+ transients are followed by neuronal death, suggesting that aberrant astrocytic Ca2+ signal results in neurotoxicity [217]. These results, however, are not universal, and other groups have not replicated these observations [218]. It has also been suggested that APOE4 dysregulates Ca2+ excitability in astrocytes by modifying membrane lipid composition. This phenomenon was observed in hippocampal slices from APOE3 and APOE4 mice, specifically in male mice [219], suggesting that the APOE genotype modulates Ca2+ fluxes in astrocytes in a lipid and sex-dependent manner. As demonstrated in primary cortical co-cultures of neurons and astrocytes, exposure to insoluble aggregates of tau failed to induce a Ca2+ response in astrocytes [36]. Unfortunately, little else is known about the effects of tau on astrocytic Ca2+, and further research is clearly warranted.
In the intact brain in vivo under physiological conditions, astrocytes show sporadic Ca2+ transients as a hallmark of astrocytic activity [220][219]. As demonstrated in cortical astrocytes of amyloid-depositing mice (APP/PS1), under pathological conditions, the frequency of spontaneous Ca2+ waves increases [18]. These same astrocytes exhibit higher resting Ca2+ levels. While overall astrocytic hyperactivity was noticed throughout the cortical tissue and not just in the vicinity of amyloid plaques, the astrocytes initiating the intracellular Ca2+ waves were located in plaque vicinity [18]. Further studies are needed to determine whether this was an effect of soluble Aβ oligomers or Aβ fibrils. Ca2+ hyperactivity in astrocytes has been associated with abnormal purinergic signaling, suggesting that reactive astrocytes release excessive amounts of ATP. This in turn activates P2Y purinoceptors mediating abnormal cytosolic Ca2+ signaling [17]. It has also been suggested that alterations in extracellular Ca2+ levels can be involved in astrocytic hyperactivity. During increased neuronal activity, extracellular Ca2+ decreases following ionotropic glutamate receptor and VGCC activation. Astrocytes sense the extracellular Ca2+ decrease and release ATP in response [221][220]. Increases in extracellular ATP trigger astrocytic Ca2+ transients and could contribute to AD-associated astrocytic hyperactivity.
4. Microglial Ca2+ as a Therapeutic Target in AD
Microglia are the major immune cells in the brain. They sense and react to alterations in brain homeostasis. They are also involved in synaptic pruning, which occurs during the first weeks of postnatal development and is critical for the maturation of neuronal networks [222][221]. Microglial activation is characterized by morphological alterations and production of pro- and anti-inflammatory mediators [222] [223]. Intracellular Ca2+ regulates microglial activation from its homeostatic resting state to a neurotoxic-activated state. Some microglial functions, including the production and release of proinflammatory factors, such as nitric oxide (NO) and certain cytokines, are Ca2+-dependent processes [224][223]. In turn, proinflammatory cytokines, tumor necrosis factor α (TNFα), interleukin 1β, and interferon γ, all increase intracellular Ca2+ levels in microglia [225[224][225][226],226,227], while anti-inflammatory cytokines decrease them [228][227].
AD has long been linked to microglial activation. Microglia surround amyloid plaques [229] [228] after they get recruited within the first days after plaque formation [230][229]. Once activated, microglia internalize and break down Aβ. Microglia activation is an early process in AD, and it has been shown to be correlated with cognitive deficits [231]. Released proinflammatory cytokines (such as IL-1β and TNF-α) by microglia might stimulate the release of proinflammatory substances by astrocytes, amplifying the inflammatory signal and its neurotoxicity [232,233][230][231]. Therefore, neurons and astrocytes in the vicinity of these plaques are likely subjected to high levels of proinflammatory mediators released by activated microglia. These mediators can cause alterations in the Ca2+ homeostasis of these cells [234][232]. Additionally, microglial cultures exposed to Aβ increase their immune response (i.e., cytokine production) and intracellular Ca2+, a process that can be blocked by the dihydropyridine nifedipine and the non-dihydropyridine L-type VGCC antagonist verapamil or diltiazem [235][233].
Observations from in vitro data have shown that intracellular Ca2+ homeostasis is impaired in activated microglia. Microglia isolated from AD brain tissue have elevated cytosolic Ca2+ levels compared to controls and exhibit reduced responsiveness to stimuli in vitro [236][234]. Additionally, mouse microglia activated by lipopolysaccharide (LPS) display increased basal Ca2+ levels and a reduced agonist-induced Ca2+ signal [224][223]. Ramified activated microglia display large intracellular Ca2+ transients in response to the damage of individual cells in their vicinity. The use of in vivo Ca2+ imaging and multiphoton microscopy has allowed the study of microglial Ca2+ dynamics in the intact living brain. Microglia display rare Ca2+ transients in their resting state, but respond with larger Ca2+ transients when activated [237][235]. Microglial Ca2+ transients are attributed to Ca2+ release from intracellular stores and are prevented by the activation of ATP receptors [237][236]. Blocking SOCE—via knocking down or knocking out STIM1/2 and Orai—reduces immune functioning, including phagocytosis, migration, and cytokine production in primary isolated murine microglia [238,239][237][238]. On the other hand, blocking RyR prevents LPS-induced neurotoxicity mediated by microglia [240][239]. Although they have not been studied in depth, it has been suggested that microglia display Ca2+ microdomains. Some observations suggest that global Ca2+ elevations in microglia trigger phagocytosis and migration, whereas local Ca2+ increases in their processes regulate acute chemotactic migration [241][240]. Taken together, elevated Ca2+ levels seem to be a hallmark of activated microglia and their regulation, a potential therapeutic target for AD therapy.
Microglia express P2X receptors, a subfamily of purinergic ionotropic receptors, located in the plasma membrane which are permeable to Ca2+, Na+, and K+ [242][241]. Overactivation of P2X receptors may lead to cell death via membrane depolarization, mitochondrial stress, and ROS production [243][242]. As measured in postmortem brain tissue from AD patients, P2X7 expression is upregulated in microglia in AD [244][243]. This same effect was shown in plaque-associated microglia in an AD mouse model and after intrahippocampal injection of Aβ42 [245][244]. It is believed that these high levels of P2X7 contribute to the enhanced inflammatory responses observed in AD [244,246,247][243][245][246]. Inhibition of P2X7 receptors has been shown to be neuroprotective as it reduces the dendritic spine loss induced by Aβ [248][247], as well as Aβ production in general [249][248]. P2X7 KO mice express reduced plaque size and improved behavioral scores [250][249], suggesting P2X7 as a potential therapeutic target in AD. Additional in vitro studies have shown that P2X7 activation leads to microglial NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome activation, which requires Ca2+ mobilization from intracellular stores [251,252][250][251]. Aβ also triggers this NLRP3 activation [253][252], and NLRP3 is highly activated in microglial cells surrounding amyloid plaques [253][252]. NLRP3 KO mice show reduced amyloid burden in the brain and have reduced memory impairment [254][253]. This confirms that P2X7 and NLRP3 could be candidate targets for AD therapeutics.
The triggering receptor expressed on myeloid cells 2 (TREM2) gene has been recently identified as a risk gene for AD [255][254]. Its low-frequency variants increase the risk of developing AD similar to the APOE4 allele. TREM2 is a transmembrane protein receptor expressed on microglia. It stimulates phagocytosis and suppresses inflammation [255][254]. TREM2 overexpression in a mouse model of AD (APP/PS1) decreased AD-related pathology and improved cognitive functions [256][255], suggesting that modeling microglial functions could be a protective target in AD. Immunotherapy using antibodies to stimulate TREM2 signaling in order to improve AD pathology is currently being developed by different groups. Stimulation with anti-TREM2 antibodies in vitro produced Ca2+ influx and extracellular signal-regulated kinase (ERK) signaling activation in human dendritic cells [257][255]. When to stimulate TREM2 to treat AD, however, is not clear, and it must be kept in mind that the use of these antibodies could alter the binding of other TREM2 ligands. Further studies will be needed to fully understand TREM2 function and its role in AD therapy.
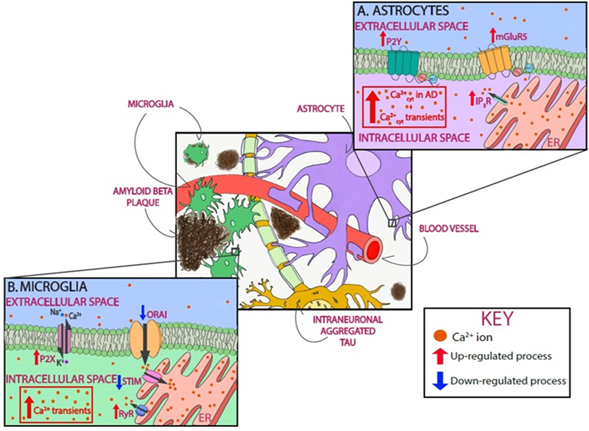
Figure 2. Astrocytic and microglial Ca2+ as a therapeutic target in AD. Schematic of glial Ca2+ cells dysregulation in the presence of AD pathology. In astrocytes, P2Y purinoceptors and glutamate metabotropic receptors mGluR5, when activated, cause Ca2+ increase by releasing Ca2+ from intracellular stores. As shown in red, all three receptors are upregulated in AD. In addition, cytosolic Ca2+ levels are increased in astrocytes, and they exhibit Ca2+ transients (A). In microglia, P2X receptors are upregulated in AD, thus leading to Ca2+ dysregulation. SOCE, with involves STIM and Orai, is also responsible for Ca2+ influx, specifically into the lumen of the endoplasmic reticulum (ER). This pathway is downregulated in AD (shown in blue). RyRs mediate Ca2+ efflux from the ER, a process that is upregulated in AD. Microglia also show Ca2+ dysregulation by showing cytosolic Ca2+ transients (B).
5. Future Directions
Current FDA-approved AD treatments target plasma Ca2+ channels, but more specific approaches are needed to target other prevalent and disrupted intracellular Ca2+ signaling pathways, such as those of the ER or mitochondria. As our knowledge in Ca2+ dysregulation in AD grows, it seems more obvious that targeting these other sources of Ca2+ dysregulation could be an effective therapeutic strategy.
A better understanding of the onset and progression of neurodegenerative diseases will facilitate rapid diagnosis and target selection, allowing for early treatment. A truly effective method for preventing or treating Alzheimer’s disease will likely involve a combination approach for targets, such as Aβ plaque clearance or soluble tau removal. Additionally, reversal of cellular processes that are disrupted by Aβ or tau accumulation (including Ca2+ dyshomeostasis), early diagnosis, and/or lifestyle changes would also be necessary for successful therapeutic intervention. Gene therapy is an emerging therapeutic strategy for the treatment of neurodegenerative disorders, including AD, particularly when traditional therapies are not responsive to well-validated genetic targets. Gene therapy has already shown efficacy in preclinical studies, utilizing different routes for gene delivery. Recently, different groups proposed gene therapy as a strategy in the battle against AD, as it is designed to focus on one specific target in affected brain regions. Several gene therapy strategies for AD have already been tested. These include acting directly on APP metabolism, neuroprotection, targeting inflammatory pathways, or modulating genes related to lipid metabolism.
References
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; E Schmechel, D.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; A Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- A Hardy, J.; A Higgins, G.; Mayford, M.; Barzilai, A.; Keller, F.; Schacher, S.; Kandel, E. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Davies, P. Selective Loss Of Central Cholinergic Neurons In Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Hypothesis on the Regulation of Cytosol Calcium Concentration and the Aging Brain. Neurobiol. Aging 1987, 8, 345–346. [Google Scholar] [CrossRef]
- Boada, M.; Antúnez, C.; López-Arrieta, J.; Galán, J.J.; Morón, F.J.; Hernández, I.; Marín, J.; Martínez-Lage, P.; Alegret, M.; Carrasco, J.M.; et al. CALHM1 P86L Polymorphism is Associated with Late-Onset Alzheimer’s Disease in a Recessive Model. J. Alzheimer’s Dis. 2010, 20, 247–251. [Google Scholar] [CrossRef]
- Tolar, M.; Keller, J.N.; Chan, S.; Mattson, M.P.; Marques, M.A.; Crutcher, K.A. Truncated Apolipoprotein E (ApoE) Causes Increased Intracellular Calcium and May Mediate ApoE Neurotoxicity. J. Neurosci. 1999, 19, 7100–7110. [Google Scholar] [CrossRef]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ornath, M.; Hudry, E.; Boivin, J.R.; Hashimoto, T.; Takeda, S.; Kuchibhotla, K.V.; Hou, S.; Lattarulo, C.R.; Belcher, A.M.; Trujillo, P.B.; et al. Soluble oligomeric amyloid-beta induces calcium dyshomeostasis that precedes synapse loss in the living mouse brain. Mol. Neurodegener. 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.-H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of Hyperactive Neurons Near Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef]
- Wang, X.; Kastanenka, K.V.; Arbel-Ornath, M.; Commins, C.; Kuzuya, A.; Lariviere, A.J.; Krafft, G.A.; Hefti, F.; Jerecic, J.; Bacskai, B.J. An acute functional screen identifies an effective antibody targeting amyloid-beta oligomers based on calcium imaging. Sci. Rep. 2018, 8, 4634. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging Clin. Exp. Res. 1989, 1, 17–34. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Abeysinghe, A.; Deshapriya, R.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V.; Imbimbo, B.P.; Logroscino, G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: The point of no return? Expert Opin. Biol. Ther. 2014, 14, 1465–1476. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neuronal injury in Alzheimer’s disease. Contributions of beta-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann. N. Y. Acad. Sci. 1994, 747, 50–76. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Pourbadie, H.G.; Naderi, N.; Mehranfard, N.; Janahmadi, M.; Khodagholi, F.; Motamedi, F. Preventing Effect of L-Type Calcium Channel Blockade on Electrophysiological Alterations in Dentate Gyrus Granule Cells Induced by Entorhinal Amyloid Pathology. PLoS ONE 2015, 10, e0117555. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, B.A.; Kennelly, S.; O’Dwyer, S.; Cregg, F.; Walsh, C.D.; Cohen, R.; Kenny, R.A.; Howard, R.; Murphy, C.; Adams, J.; et al. NILVAD protocol: A European multicentre double-blind placebo-controlled trial of nilvadipine in mild-to-moderate Alzheimer’s disease. BMJ Open 2014, 4, e006364. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, B.A.; Segurado, R.; Kennelly, S.; Rikkert, M.G.M.O.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Minster, R.L.; Demirci, F.Y.; DeKosky, S.T.; Kamboh, M.I. No association betweenCALHM1variation and risk of Alzheimer disease. Hum. Mutat. 2009, 30, E566–E569. [Google Scholar] [CrossRef]
- Tan, E.; Ho, P.; Cheng, S.; Yih, Y.; Li, H.; Fook-Chong, S.; Lee, W.; Zhao, Y. CALHM1 variant is not associated with Alzheimer’s disease among Asians. Neurobiol. Aging 2011, 32, 546.e11–546.e12. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Chang, E.H.; Frattini, S.A.; Zhao, H.; Chandakkar, P.; Adrien, L.; Strohl, J.J.; Gibson, E.L.; Ohmoto, M.; Matsumoto, I.; et al. CALHM1 deficiency impairs cerebral neuron activity and memory flexibility in mice. Sci. Rep. 2016, 6, 24250. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.; Rego, A.C. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schroder, U.H.; Fandrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR 2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef]
- Esclaire, F.; Lesort, M.; Blanchard, C.; Hugon, J. Glutamate toxicity enhances tau gene expression in neuronal cultures. J. Neurosci. Res. 1997, 49, 309–318. [Google Scholar] [CrossRef]
- Sindou, P.; Lesort, M.; Couratier, P.; Yardin, C.; Esclaire, F.; Hugon, J. Glutamate increases tau phosphorylation in primary neuronal cultures from fetal rat cerebral cortex. Brain Res. 1994, 646, 124–128. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Tuo, Q.-Z.; Liuyang, Z.-Y.; Xie, A.-J.; Feng, X.-L.; Yan, X.; Qiu, M.; Li, S.; Wang, X.-L.; Cao, F.-Y.; et al. Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. 2016, 7, e2449. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Failures and successes of NMDA receptor antagonists: Molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s Disease: An Updated Systematic Review and Meta-analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.P.; Clarke, J.R.; Ledo, J.H.; Ribeiro, F.C.; Costa, C.V.; Melo, H.M.; Mota-Sales, A.P.; Saraiva, L.M.; Klein, W.L.; Sebollela, A. et al.; et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 2013, 33, 9626–9634. [Google Scholar] [CrossRef]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against β-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Wevers, A.; Monteggia, L.; Nowacki, S.; Bloch, W.; Schutz, U.; Lindstrom, J.; Pereira, E.F.R.; Eisenberg, H.; Giacobini, E.; De Vos, R.A.I.; et al. Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer’s disease: Histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. Eur. J. Neurosci. 1999, 11, 2551–2565. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Rogers, J.T.; Greig, N.H.; Sambamurti, K. Rationale for the Development of Cholinesterase Inhibitors as Anti- Alzheimer Agents. Curr. Pharm. Des. 2004, 10, 3111–3119. [Google Scholar] [CrossRef]
- Herholz, K. Acetylcholine esterase activity in mild cognitive impairment and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 25–29. [Google Scholar] [CrossRef]
- Patel, L.; Grossberg, G.T. Combination Therapy for Alzheimerʼs Disease. Drugs Aging 2011, 28, 539–546. [Google Scholar] [CrossRef]
- Gómez-Ramos, A.; Díaz-Hernández, M.; Rubio, A.; Miras-Portugal, M.T.; Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 2008, 37, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J.A.; Johnson, G.V.W. Transient Increases in Intracellular Calcium Result in Prolonged Site-selective Increases in Tau Phosphorylation through a Glycogen Synthase Kinase 3β-dependent Pathway. J. Biol. Chem. 1999, 274, 21395–21401. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—Moving from amyloid-beta to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef]
- Kang, S.; Son, S.M.; Baik, S.H.; Yang, J.; Mook-Jung, I. Autophagy-Mediated Secretory Pathway is Responsible for Both Normal and Pathological Tau in Neurons. J. Alzheimer’s Dis. 2019, 70, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Devos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef]
- Friedhoff, P.; Von Bergen, M.; Mandelkow, E.-M.; Davies, P. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc. Nat. Acad. Sci. USA 1998, 95, 15712–15717. [Google Scholar] [CrossRef]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.-P.; Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; De Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 1–31. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Palop, J.J. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Nat. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with Aducanumab Restores Calcium Homeostasis in Tg2576 Mice. J. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPHIE, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Nat. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-In Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.-O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 Receptor-Mediated Ca2+ Signaling Alleviates Mutant Presenilin-Linked Familial Alzheimer’s Disease Pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nat. Cell Biol. 1999, 398, 522–525. [Google Scholar] [CrossRef]
- A Saura, C.; Choi, S.-Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Rao, B.S.S.; Chattarji, S.; Kelleher, R.J.; Kandel, E.R.; Duff, K.; et al. Loss of Presenilin Function Causes Impairments of Memory and Synaptic Plasticity Followed by Age-Dependent Neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef]
- Watanabe, H.; Xia, D.; Kanekiyo, T.; Kelleher, R.J.; Shen, J. Familial Frontotemporal Dementia-Associated Presenilin-1 c.548G>T Mutation Causes Decreased mRNA Expression and Reduced Presenilin Function in Knock-In Mice. J. Neurosci. 2012, 32, 5085–5096. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Zhang, J.; Kholodenko, D.; Citron, M.; Podlisny, M.B.; Teplow, D.B.; Haass, C.; Seubert, P.; Koo, E.H.; Selkoe, D.J. Enhanced Production and Oligomerization of the 42-residue Amyloid β-Protein by Chinese Hamster Ovary Cells Stably Expressing Mutant Presenilins. J. Biol. Chem. 1997, 272, 7977–7982. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Shen, J.; Iii, R.J.K. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Nat. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.E.; Gibson, E.A.; Palmer, A.E. Using a genetically targeted sensor to investigate the role of presenilin-1 in ER Ca2+ levels and dynamics. Mol. Biosyst. 2010, 6, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Mak, D.-O.D.; Kang, D.E.; Foskett, J.K. Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+ Leak Channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 Is an ER Ca2+ Load-Activated Ca2+ Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef]
- Bruno, A.M.; Huang, J.Y.; Bennett, D.A.; Marr, R.A.; Hastings, M.L.; Stutzmann, G.E. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1001.e1–1001.e6. [Google Scholar] [CrossRef]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 Mutations Increase Levels of Ryanodine Receptors and Calcium Release in PC12 Cells and Cortical Neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef]
- Rybalchenko, V.; Hwang, S.-Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef]
- Wu, B.; Yamaguchi, H.; Lai, F.A.; Shen, J. Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc. Nat. Acad. Sci. USA 2013, 110, 15091–15096. [Google Scholar] [CrossRef]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, I.; Györke, S.; Zahradníková, A. Calcium Activation of Ryanodine Receptor Channels—Reconciling RyR Gating Models with Tetrameric Channel Structure. J. Gen. Physiol. 2005, 126, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-β-(1-42) Increases Ryanodine Receptor-3 Expression and Function in Neurons of TgCRND8 Mice. J. Biol. Chem. 2006, 281, 38440–38447. [Google Scholar] [CrossRef]
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.; Peng, Y.; Eckenhoff, M.F.; Wei, H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett. 2012, 516, 274–279. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, B.; Liu, C.; Liang, G.; Liu, W.; Pickup, S.; Meng, Q.; Tian, Y.; Li, S.; Eckenhoff, M.F.; et al. Long-term Dantrolene Treatment Reduced Intraneuronal Amyloid in Aged Alzheimer Triple Transgenic Mice. Alzheimer Dis. Assoc. Disord. 2015, 29, 184–191. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Pani, B.; Singh, B.B. Neurological and Motor Disorders: Neuronal Store-Operated Ca2+ Signaling: An Overview and Its Function. Atherosclerosis 2017, 993, 535–556. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. The Physiological Function of Store-operated Calcium Entry. Neurochem. Res. 2011, 36, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef]
- Smith, I.; Boyle, J.; Vaughan, P.; Pearson, H.; Cowburn, R.; Peers, C. Ca2+ Stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer’s disease presenilin-1 mutation. Brain Res. 2002, 949, 105–111. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca2+ Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+ Remodeling. Front. Cell. Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-Mediated Modulation of Capacitative Calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.D.; Saito, T.; Saido, T.C.; Bezprozvanny, I.B. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef]
- Tsukamoto, A.; Kaneko, Y. Thapsigargin, a Ca2+-ATPase inhibitor, depletes the intracellular Ca2+ pool and induces apoptosis in human hepatoma cells. Cell Biol. Int. 1993, 17, 969–970. [Google Scholar] [CrossRef]
- Soboloff, J.; Berger, S.A. Sustained ER Ca2+ Depletion Suppresses Protein Synthesis and Induces Activation-enhanced Cell Death in Mast Cells. J. Biol. Chem. 2002, 277, 13812–13820. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Pchitskaya, E.; Speshilova, A.B.; Alexandrov, S.; Zhang, H.; Vlasova, O.; Bezprozvanny, I.B. STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 2015, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, E.; Zhou, Q.; Li, S.; Wang, X.; Liu, Y.; Wang, L.; Sun, D.; Ye, J.; Gao, Y.; et al. TRPC1 Null Exacerbates Memory Deficit and Apoptosis Induced by Amyloid-beta. J. Alzheimer’s Dis. JAD 2018, 63, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Werth, J.L.; A Thayer, S. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 1994, 14, 348–356. [Google Scholar] [CrossRef]
- Billups, B.; Forsythe, I.D. Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian Central Synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar] [CrossRef]
- Wescott, A.P.; Kao, J.P.Y.; Lederer, W.J.; Boyman, L. Voltage-energized calcium-sensitive ATP production by mitochondria. Nat. Metab. 2019, 1, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; A Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nat. Cell Biol. 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Doonan, P.J.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 Is an Essential Gatekeeper for MCU-Mediated Mitochondrial Ca2+ Uptake that Regulates Cell Survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Reane, D.V.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 Finely Tune the Mitochondrial Ca2+ Uniporter by Exerting Opposite Effects on MCU Activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 Controls Both the Threshold and Cooperative Activation of the Mitochondrial Ca2+ Uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress–mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Nat. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V.; et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nat. Cell Biol. 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA Copy Numbers in Pyramidal Neurons are Decreased and Mitochondrial Biogenesis Transcriptome Signaling is Disrupted in Alzheimer’s Disease Hippocampi. J. Alzheimer’s Dis. 2014, 40, 319–330. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Wang, J.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 2006, 96, 825–832. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 Tau Fragment Targets Neuronal Mitochondria at AD Synapses: Possible Implications for Neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodríguez, M.; García-Durillo, M.; Villalobos, C.; Núñez, L. Aging Enables Ca2+ Overload and Apoptosis Induced by Amyloid-beta Oligomers in Rat Hippocampal Neurons: Neuroprotection by Non-Steroidal Anti-Inflammatory Drugs and R-Flurbiprofen in Aging Neurons. J. Alzheimer’s Dis. JAD 2016, 54, 207–221. [Google Scholar] [CrossRef]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Yan, Y.; Gong, K.; Ma, T.; Zhang, L.; Zhao, N.; Zhang, X.; Tang, P.; Gong, Y. Protective effect of edaravone against Alzheimer’s disease-relevant insults in neuroblastoma N2a cells. Neurosci. Lett. 2012, 531, 160–165. [Google Scholar] [CrossRef]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid β-Peptide Promotes Permeability Transition Pore in Brain Mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Skoog, I.; Östling, S.; Kern, J.; Börjesson-Hanson, A. Does low-dose acetylsalicylic acid prevent cognitive decline in women with high cardiovascular risk? A 5-year follow-up of a non-demented population-based cohort of Swedish elderly women. BMJ Open 2012, 2, e001288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis From Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.; Meyer, P.-F. Author response: INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2020, 94, 594. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Calvo-Rodriguez, M.; Caballero, E.; Garcia-Durillo, M.; Nunez, L.; Villalobos, C. Is it All Said for NSAIDs in Alzheimer’s Disease? Role of Mitochondrial Calcium Uptake. Curr. Alzheimer Res. 2018, 15, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Gutknecht, J. Salicylates and proton transport through lipid bilayer membranes: A model for salicylate-induced uncoupling and swelling in mitochondria. J. Membr. Biol. 1990, 115, 253–260. [Google Scholar] [CrossRef]
- Núñez, L.; Valero, R.A.; Senovilla, L.; Sanz-Blasco, S.; García-Sancho, J.; Villalobos, C. Cell proliferation depends on mitochondrial Ca2+ uptake: Inhibition by salicylate. J. Physiol. 2006, 571, 57–73. [Google Scholar] [CrossRef]
- Calvo-Rodríguez, M.; Sanz-Blasco, S.; Caballero, E.; Villalobos, C.; Núñez, L. Susceptibility to excitotoxicity in aged hippocampal cultures and neuroprotection by non-steroidal anti-inflammatory drugs: Role of mitochondrial calcium. J. Neurochem. 2015, 132, 403–417. [Google Scholar] [CrossRef]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons Against Glutamate Excitotoxicity. Mol. Neurobiol. 2019, 56, 2244–2255. [Google Scholar] [CrossRef]
- Dubey, M.; Chaudhury, P.; Kabiru, H.; Shea, T.B. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: Neurofilaments attenuate tau-mediated neurite instability. Cell Motil. Cytoskelet. 2008, 65, 89–99. [Google Scholar] [CrossRef]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V. Caspase-cleaved Tau Expression Induces Mitochondrial Dysfunction in Immortalized Cortical Neurons. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [PubMed]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- García-Pérez, C.; Hajnóczky, G.; Csordás, G. Physical Coupling Supports the Local Ca2+ Transfer between Sarcoplasmic Reticulum Subdomains and the Mitochondria in Heart Muscle. J. Biol. Chem. 2008, 283, 32771–32780. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Nat. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef]
- Perreault, S.; Bousquet, O.; Lauzon, M.; Paiement, J.; Leclerc, N. Increased Association Between Rough Endoplasmic Reticulum Membranes and Mitochondria in Transgenic Mice That Express P301L Tau. J. Neuropathol. Exp. Neurol. 2009, 68, 503–514. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Castillo, M.D.C.L.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.F. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Nat. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Smith, M.A.; Perry, G.H.; Aliev, G. Mitochondrial failures in Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2004, 19, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Ortiz, J.M.P.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Szeto, H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Kandimalla, R. Mitochondria-targeted small molecule SS31: A potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 1597. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Reddy, A.P. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Morris, J.K. New Therapeutics to Modulate Mitochondrial Function in Neurodegenerative Disorders. Curr. Pharm. Des. 2017, 23, 731–752. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca2+ homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; A Levey, S.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef]
- Bach, G.; Chen, C.-S.; E Pagano, R. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin. Chim. Acta 1999, 280, 173–179. [Google Scholar] [CrossRef]
- Soyombo, A.A.; Tjon-Kon-Sang, S.; Rbaibi, Y.; Bashllari, E.; Bisceglia, J.; Muallem, S.; Kiselyov, K. TRP-ML1 Regulates Lysosomal pH and Acidic Lysosomal Lipid Hydrolytic Activity. J. Biol. Chem. 2006, 281, 7294–7301. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Lee, J.-H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.S.; Coffey, E.E.; Kompella, U.B.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Neely, K.M.; Green, K.N.; LaFerla, F.M. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J. Neurosci. 2011, 31, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Wolfe, D.M.; Lee, J.-H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Recent advances in (patho)physiology of astroglia. Acta Pharmacol. Sin. 2010, 31, 1044–1054. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Guerra-Gomes, S.; Sousa, N.; Pinto, L.; Oliveira, J.F. Functional Roles of Astrocyte Calcium Elevations: From Synapses to Behavior. Front. Cell. Neurosci. 2018, 11, 427. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Iyer, A.; Ronco, V.; A Grolla, A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sanchez-Gomez, M.V.; Cavaliere, F.; Rodriguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef]
- Toivari, E.; Manninen, T.; Nahata, A.K.; Jalonen, T.O.; Linne, M.-L. Effects of Transmitters and Amyloid-Beta Peptide on Calcium Signals in Rat Cortical Astrocytes: Fura-2AM Measurements and Stochastic Model Simulations. PLoS ONE 2011, 6, e17914. [Google Scholar] [CrossRef]
- Larramona-Arcas, R.; González-Arias, C.; Perea, G.; Gutiérrez, A.; Vitorica, J.; García-Barrera, T.; Gómez-Ariza, J.L.; Pascua-Maestro, R.; Ganfornina, M.D.; Kara, E.; et al. Sex-dependent calcium hyperactivity due to lysosomal-related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Mol. Neurodegener. 2020, 15, 1–23. [Google Scholar] [CrossRef]
- Hirase, H.; Qian, L.; Barthó, P.; Buzsáki, G. Calcium Dynamics of Cortical Astrocytic Networks In Vivo. PLoS Biol. 2004, 2, e96. [Google Scholar] [CrossRef]
- Torres, A.; Wang, F.; Xu, Q.; Fujita, T.; Dobrowolski, R.; Willecke, K.; Takano, T.; Nedergaard, M. Extracellular Ca2+ Acts as a Mediator of Communication from Neurons to Glia. Sci. Signal. 2012, 5, ra8. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Tichauer, J.E.; Eugenín, J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J. Neurochem. 2010, 112, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.-K.; Kettenmann, H. Elevation of Basal Intracellular Calcium as a Central Element in the Activation of Brain Macrophages (Microglia): Suppression of Receptor-Evoked Calcium Signaling and Control of Release Function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, S.; Choi, H.B.; Kim, S.U.; McLarnon, J.G. Interferon-? acutely induces calcium influx in human microglia. J. Neurosci. Res. 2002, 69, 607–613. [Google Scholar] [CrossRef]
- Goghari, V.; Franciosi, S.; Kim, S.U.; Lee, Y.B.; McLarnon, J.G. Acute application of interleukin-1beta induces Ca2+ responses in human microglia. Neurosci. Lett. 2000, 281, 83–86. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Franciosi, S.; Wang, X.; Bae, J.; Choi, H.; Kim, S. Acute actions of tumor necrosis factor-α on intracellular Ca2+ and K+ currents in human microglia. Neuroscience 2001, 104, 1175–1184. [Google Scholar] [CrossRef]
- Turovskaya, M.V.; Turovsky, E.A.; Zinchenko, V.P.; Levin, S.G.; Godukhin, O.V. Interleukin-10 modulates [Ca2+]i response induced by repeated NMDA receptor activation with brief hypoxia through inhibition of InsP3-sensitive internal stores in hippocampal neurons. Neurosci. Lett. 2012, 516, 151–155. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.; Akiyama, H.; Zhu, S.; Selkoe, D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989, 24, 173–182. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Sun, M.-Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front. Pharmacol. 2012, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Brawek, B.; Garaschuk, O. Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res. 2014, 357, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Silei, V.; Fabrizi, C.; Venturini, G.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Lauro, G.M. Activation of microglial cells by PrP and beta-amyloid fragments raises intracellular calcium through L-type voltage sensitive calcium channels. Brain Res. 1999, 818, 168–170. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Choi, H.B.; Lue, L.-F.; Walker, D.G.; Kim, S.U. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J. Neurosci. Res. 2005, 81, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1014–1024. [Google Scholar] [CrossRef]
- Heo, D.K.; Lim, H.M.; Nam, J.H.; Lee, M.G.; Kim, J.Y. Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 2015, 27, 177–186. [Google Scholar] [CrossRef]
- Michaelis, M.; Nieswandt, B.; Stegner, D.; Eilers, J.; Kraft, R. STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 2015, 63, 652–663. [Google Scholar] [CrossRef]
- Klegeris, A.; Choi, H.B.; McLarnon, J.G.; McGeer, P.L. Functional ryanodine receptors are expressed by human microglia and THP-1 cells: Their possible involvement in modulation of neurotoxicity. J. Neurosci. Res. 2007, 85, 2207–2215. [Google Scholar] [CrossRef]
- Lim, H.M.; Woon, H.; Han, J.W.; Baba, Y.; Kurosaki, T.; Lee, M.G.; Kim, J.Y. UDP-Induced Phagocytosis and ATP-Stimulated Chemotactic Migration Are Impaired in STIM1(-/-) Microglia In Vitro and In Vivo. Mediat. Inflamm. 2017, 2017, 8158514. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Chan, C.M.; Loesch, A.; Unwin, R.; Burnstock, G. Induction of proliferation and apoptotic cell death via P2Y and P2X receptors, respectively, in rat glomerular mesangial cells. Kidney Int. 2000, 57, 949–958. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G.; Ryu, J.K.; Walker, D.G.; Choi, H.B. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R. P2 × 7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid {beta} requires P2 × 7 receptor expression. J. Immunol. Res. 2009, 182, 4378–4385. [Google Scholar]
- Illes, P.; Rubini, P.; Huang, L.; Tang, Y. The P2 × 7 receptor: A new therapeutic target in Alzheimer’s disease. Expert Opin. 2019, 23, 165–176. [Google Scholar]
- Jana, M.K.; Cappai, R.; Ciccotosto, G.D. Oligomeric Amyloid-beta Toxicity Can Be Inhibited by Blocking Its Cellular Binding in Cortical Neuronal Cultures with Addition of the Triphenylmethane Dye Brilliant Blue, G. ACS Chem. Biol. 2016, 7, 1141–1147. [Google Scholar]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; Leon-Otegui, M.; Hontecillas-Prieto, L.; Del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In vivo P2 × 7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3beta and secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef]
- Martin, E.; Amar, M.; Dalle, C.; Youssef, I.; Boucher, C.; Le Duigou, C.; Bruckner, M.; Prigent, A.; Sazdovitch, V.; Halle, A.; et al. New role of P2 × 7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 2019, 24, 108–125. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nat. Cell Biol. 2012, 492, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Nat. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher-Moser, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Tan, M.-S.; Gu, L.-Z.; Wang, H.-F.; Ding, Z.-Z.; Zhang, Y.-D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A Dap12-Mediated Pathway Regulates Expression of Cc Chemokine Receptor 7 and Maturation of Human Dendritic Cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [PubMed]