The gastrointestinal (GI) mucosa is among the most complex systems in the body. It has a diverse commensal microbiome challenged continuously by food and microbial components while delivering essential nutrients and defending against pathogens. For these reasons, regulatory cells and receptors are likely to play a central role in maintaining the gut mucosal homeostasis. Recent lessons from cancer immunotherapy point out the critical role of the B7 negative co-stimulator PD-L1 in mucosal homeostasis. In this entreviewy, we summarize the current knowledge supporting the critical role of PD-L1 in gastrointestinal mucosal tolerance and how abnormalities in its expression and signaling contribute to gut inflammation and cancers. Abnormal expression of PD-L1 and/or the PD-1/PD-L1 signaling pathways have been observed in the pathology of the GI tract. We also discuss the current gap in our knowledge with regards to PD-L1 signaling in the GI tract under homeostasis and pathology. Finally, we summarize the current understanding of how this pathway is currently targeted to develop novel therapeutic approaches.
- PD-L1
- gastrointestinal inflammation
- homeostasis
- inflammatory bowel disease
1. Introduction
The intestinal tract is a complicated and well-orchestrated system, comprising different types of cells, including dendritic cells (DC), macrophages, T cells, B cells, innate lymphoid cells (ILCs), as well as epithelial, mesenchymal, and endothelial cells. The tightly regulated interactions between those cells segregate commensal microbes and maintain a balance to ensure tissue health and regeneration. The continuous crosstalk between gut microbiota and the host gastrointestinal (GI) tract supports a well-balanced relationship between gut microbes and the host’s immune system [1]. One of the major processes in intestinal homeostasis, which controls local inflammatory responses in the gut, is mucosal tolerance [2].
Over the last decade, signaling through the B7 negative co-stimulator, PD-L1, has emerged as a key mechanism for the mucosal tolerance in the gut [3]. The engagement of PD-1 on T cells by PD-L1 inhibits the activation and proliferation of effector T cells [4], inducing those producing inflammatory IFN-g and IL-17A cytokines. Furthermore, PD-L1 has been shown to induce regulatory T cells (Tregs) [5,6][5][6]. PD-L1 expression is normally upregulated during inflammation to prevent overt tissue damage [3]. The importance of this molecule in regulating immune responses in the intestinal tract and maintaining tolerance was established in animal models using PD-L1 and/or PD-1 knockouts and transgenic mice [7]. Abnormal expression of PD-L1 and/or PD-L1/PD-1 signaling have been observed in Inflammatory Bowel Diseases (IBD) [8,9,10,11,12,13][8][9][10][11][12][13], Helicobacter pylori chronic infection [14,15][14][15] celiac disease [16], and GI cancers [17,18,19,20][17][18][19][20]. Studies in preclinical models and clinical data suggest that PD-L1 may serve as a prognostic marker and therapeutic target in several GI chronic inflammatory diseases and cancer. PD-1 blockade has been shown to reinvigorate exhausted T cells, providing enhanced anti-tumor responses [21]. These observations have led to the development of an anti-cancer immune checkpoint therapy targeting PD-L1/PD-1 signaling [5,22][5][22]. Importantly, gastrointestinal adverse events following immune checkpoint blockade in cancer patients point out the importance of the baseline expression of these molecules in gut homeostasis [23,24][23][24]. At the same time, targeting immune checkpoints in given subgroups of IBD patients with an abnormality in PD-L1 signaling was proposed as a target for the development of better personalized therapeutic approaches [25].
2. PD-L1 in Gut Homeostasis
Over the last decade, studies have shown that PD-L1/PD-1 signaling is critical to regulating both innate and adaptive immune responses in gut mucosa under homeostasis. The expression of PD-L1 by non-hematopoietic cells is suggested to regulate self-reactive T cells or B cells and inflammatory responses in the gut and its associated gut-associated lymphoid tissue (GALTs) [5,34][5][26]. PD-L1-positive stromal cells were reported to inhibit granzyme B production in CD8+ T effector cells in vitro [35][27]. PD-L1-mediated signaling on the mesenchymal component of the mucosal lamina propria has been shown to suppress IFN-γ and IL-17A producing T helper (Th) cell responses in the colon [12,36,37][12][28][29]. In general, PD-L1 has been shown to regulate the development, maintenance, and function of inducible Foxp3+ Treg in vitro and in vivo [6].
The basal level of PD-L1 expression was detected under homeostasis in both the upper and lower gastrointestinal tract. For example, using immunohistochemistry on the paraffin-embedded tissue, Mezache et al. demonstrated a high basal expression of PD-L1 by epithelial cells within the gastric gland [38][30]. We reported the expression of PD-L1 on both mRNA and protein levels within mucosal lamina propria in the small intestine and colon [11]. In particular, the constitutive expression of PD-L1 has been reported for small intestinal and colonic epithelial cells and suggested to be critical in the epithelial mediated control over proliferation of the CD4+ and CD8+ T cells [8]. Our group observed that within the colonic mucosa, CD90+ mesenchymal cells were the major cells expressing surface PD-L1 in the normal human colon [11], and suggested the role of that molecule in maintaining immune tolerance in the intestinal tract [12,36][12][28]. A similar observation was made by us for murine colonic mucosa, where activated mesenchymal cells, known as α-SMA+ myofibroblasts, were the major cells expressing PD-L1 [36][28].
The expression of PD-L1 and its receptor PD-1 was found to be significant in the germinal centers of the Peyer’s Patches (PPs). PD-L1 was found to be abundantly expressed by dendritic cells, macrophages, B cells, and upregulated in plasma cells, while T cells were major expressors of PD-1 [39][31]. Additionally, PD-L1/PD-1-mediated signaling was suggested to be critical in the regulation of the plasma cell responses. Lack of the PD-L1 receptor, PD-1, in mice leads to an excess of T follicular helper (Tfh) cells with altered phenotypes resulting in a dysregulated selection of IgA precursor cells in the germinal centers of PPs [40][32]. This signaling pathway was shown to be critical to the maintenance of a healthy microbiome through the regulation of the IgA selection [39,40][31][32]. Indeed, PD-1KO mice have a dramatically alteration in microbiota such as increases in Erysipelotrichaceae, Prevotellaceae, and Alcaligenaceae, while Bifidobacterium and Bacteroidaceae were undetectable. PD-1 deficiency was shown to affect the selection of IgAs in the gut that resulted in the reduced bacteria-binding capacity of IgA [39][31]. Furthermore, blocking of PD-L1 during activation of T cells with Staphylococcus aureus was reported to reduce the induction of Foxp3+CD25+CD127low T cells [41][33]. Additionally, while less studied, PD-L1:B7-1-mediated interactions between the non-hematopoietic components of the colonic mucosa and macrophages were suggested to control intestinal inflammatory responses [42][34]. Recent evidence suggests that PD-L1 also contributes to the regulation of innate lymphoid cell 2 (ILC2), ILC3, and small intestinal lamina propria lymphoid tissue inducer (LTi) cell function [43] [35] Indeed, PD-L1 deficiency on ILC2s disrupts Th2 polarization and cytokine production, leading to delayed worm expulsion during infection with the gastrointestinal helminth Nippostrongylus brasiliensis [44] [36].
The critical role of gut mucosal PD-L1 in tolerogenic responses under homeostasis was pointed out by Reiynoso et al. [45][37]. Using an iFABP-tOVA transgenic mouse model, in which OVA was expressed as a self-Ag throughout the small intestine and an adoptive transfer of naive OVA-specific CD8+ T cells, it was demonstrated that abolishing PD-L1/PD-1 signaling resulted in a break of intestinal tolerance to intestinal self-Ag and induced CD8+ T cell-mediated autoimmune enteritis [45][37].
Despite advances in our understanding of the contribution of PD-L1 to gut mucosal tolerance, significant gaps remain in our knowledge of how this molecule’s expression is regulated during gut homeostasis. We previously reported that signaling through MyD88-dependent TLRs is required to maintain PD-L1 expression on mesenchymal stromal cells in the normal colonic mucosa [36][28]. The expression of PD-L1 is upregulated by several inflammatory cytokines and growth factors. Indeed, an increase in PD-L1 by IFN-γ was reported in several studies on macrophages, dendritic cells, and lymphocytes [46,47,48,49[38][39][40][41]] as well as on mesenchymal and epithelial cells [8,11][8][11]. Importantly, this cytokine was earlier reported to be produced by lymphocytes under homeostasis, in particular in duodenum [50][42]. TGF-β, which plays a critical role in the colonic mucosal tolerance [51][43], has also been suggested to be important in the expression of PD-L1 by DCs in the colon [52][44]. Thus, while further studies are needed to understand the mechanisms contributing to the regulation of PD-L1 expression and its interaction with other positive and negative B7 co-stimulators, it is clear that the maintenance of the basal level of PD-L1 is critical to gut homeostasis.
3. PD-L1/PD-1 as a Potential Therapeutic Target in Gut Chronic Inflammatory Diseases: Lesson Learned from Immune Checkpoint Therapy of Solid Cancers
PD-L1 expression on tumor cells and PD-L1 and PD-1 expression on immune cells are key mechanisms by which tumor cells escape anti-tumor immune surveillance via the suppression of the anti-tumor effector T cell response [22]. PD-L1 expression is significantly increased in several solid tumors, including microsatellite instability (MSI) colon cancer [102,103][45][46]. At the same time, in some colorectal cancers, PD-L1 is only expressed on tumor-infiltrating cells and is rarely found on tumor cells [103][46]. This places great significance on the role of PD-L1 in the tumor microenvironment. Indeed, stromal PD-L1 was associated with less aggressive tumor progression in colon cancer patients and better survival [18].
Nowadays, immune checkpoint inhibitors have emerged as a remarkable treatment option for diverse cancer types. However, a significant number of patients on checkpoint inhibitors develop immune-related adverse events (irAEs), affecting a wide variety of organs [104][47]. The most common adverse events of checkpoint blockade are gastrointestinal-adverse events [105,106][48][49]. Immune checkpoint-induced colitis (ICI colitis) is considered a distinct form of colitis with an acute onset and rapid progression, which leads to potential complications, including bowel perforation [107][50]. This type of side effect usually develops within the first few weeks or months after start of treatment but can occur at any time, including after stopping immune checkpoint blockade therapy [108][51].
A dominant feature of ICI-active colitis is the neutrophilic infiltration, crypt microabscesses, and prominent crypt epithelial cell apoptosis. However, a lymphocytic colitis-like pattern with increased intraepithelial lymphocytes was also observed [107,109][50][52]. Remarkably, PD-L1/PD-1 expression depended on ICI colitis recurring as a chronic histological manifestation consisting of basal lymphoplasmacytosis, crypt architectural irregularity, and a case of Paneth cell metaplasia, all reminiscent of IBD [110][53]. Also similar to IBD, ICI colitis is characterized by immunological changes, such as (1) CD4+ T cells predominant mucosal lamina propria infiltrate, and Th1/Th17 upregulation with normal Th2 expression (observed as well in CD); (2) elevated expression TNF-α and TNFR-like proteins (observed in both, CD and UC); and (3) mucosal abnormality in the expression of Foxp3 and IL-10 (observed in both, CD and UC) [105][48].
Recent data demonstrated the critical role of microbiota in predicting the development of ICI colitis and impacting the expression of PD-L1 and/or its receptor PD-1 [105][48]. While further studies are required to evaluate the safety of the use of probiotics in cancer patients, the use of probiotics may be helpful to render tumors sensitive to the PD-L1/PD-1 immune checkpoint therapy through the modulation of expression of these molecules. However, there is a gap in our knowledge when it comes to understanding the impact of gut microbiota on PD-L1 expression and regulation of PD-L1/PD-1 signaling. However, recent studies demonstrated that the commensal enteric strain of Escherichia coli K12 upregulated PD-L1 in IFN-γ sensitized colonic cancer epithelial cell lines, and that this process was NF-κB-dependent [111][54]. Interestingly, the same study showed that E. coli strain L20, which was isolated from a CD patient, did not affect PD-L1 expression. Furthermore, another commensal strain, Enterococcus faecalis, even significantly inhibited PD-L1 expression [111][54]. Other groups recently demonstrated that commensal strain of Pediococcus pentosaceus sp. derived extracellular membrane vesicles upregulated PD-L1 on bone marrow-derived macrophages and bone marrow progenitor cells in culture and induced recruitment of PD-L1-expressing myeloid cells to the wound site in vivo [112][55]. Furthermore, the probiotic strain of Bifidobacterium infantis has been shown to upregulate PD-L1 in a model of acute murine colitis [113][56]. Thus, a further understanding of the interactions between the gut microbiome and PD-1 ligands is needed as a potential avenue to modulate these immune checkpoints in IBD and ICI.
Patients with autoimmune disease or immune-related predisposition, such as IBD, have been excluded from most immune checkpoint clinical trials because of the potential for increased toxicity [114][57]. Indeed, gastrointestinal adverse events, such as diarrhea, in patients with underlying IBD who received immune checkpoint inhibitors, are at high risk [115][58]. Notwithstanding, treatment with anti-PD-1 antibodies in melanoma patients induce relatively frequent immune toxicities in patients with baseline autoimmunity/chronic inflammatory diseases or prior immune-related adverse events. However, recurrence of ICI colitis was rare even in patients with preexisting autoimmune diseases that were treated with TNF-α inhibitors [68][59]. Therefore, the benefits versus risks of the PD-L1/PD-1 immunotherapy in this population should be considered [68,114][59][57]. Together, these data suggest that preexisting autoimmune conditions are not an absolute contraindication to the immune checkpoint inhibitors therapy, but need a thoughtful approach. Thus, lessons learned from immune checkpoint therapy as well IBD preclinical models suggest that the effectiveness of at least PD-L1 blockade or its “supplementation” may be considered as a therapeutic approach in patients with chronic inflammatory disease of the gut where abnormality in PD-L1/PD-1 signals were reported. Therefore, understanding the potential of these molecules as a therapeutic target in several types of IBD and celiac disease warrants further investigation.
4. Conclusions
While dramatic progress has been made in the understanding of the role of PD-L1, in the maintenance of gut homeostasis and inflammatory disease, it is far from complete. It is clearly established that PD-L1 is critical to gut mucosal tolerance. Abnormality in PD-L1 expression and/or signaling was observed in gut chronic inflammatory diseases such as Crohn’s disease (CD), ulcerative colitis (UC), celiac disease, as well chronic infections such as Helicobacter pylori (Figure 1). However, how and why these abnormalities occur and their impact on disease development and progression requires further investigation. Furthermore, a more extensive understanding of the PD-L1 intrinsic signaling in cells implicated with the discussed GI pathologies is warranted. The impact of the membrane associated versus soluble form of PD-L1 in gut health and diseases warrant further investigation. With this consideration, the role of commensal and dysbiotic microbiota in the regulation of these immune checkpoints should be further investigated. Finally, the mechanisms contributing to the regulation of PD-L1 expression and its interaction with other positive and negative B7 co-stimulators during gut homeostasis and chronic inflammatory diseases are only at the beginning of understanding. Therefore, further studies are needed to address these key gaps in the field and provide a scientific basis for the design of novel PD-L1 targeting approaches for the treatment of chronic inflammatory disease of the gut.
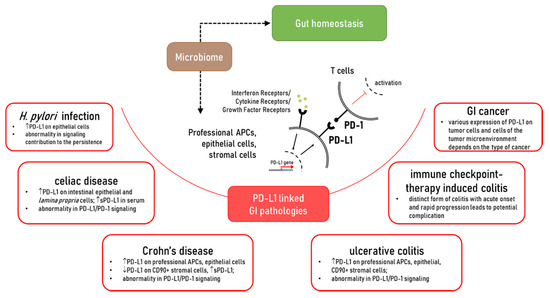
Figure 1. The role of PD-L1 in the gut homeostasis and pathologies which affected PD-L1 expression and/or PD-L1/PD-1 signaling.
References
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, 338.
- Chassaing, B.; Kumar, M.; Baker, M.T.; Singh, V.; Vijay-Kumar, M. Mammalian gut immunity. Biomed. J. 2014, 37, 246–258.
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704.
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201.
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299.
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029.
- Zamani, M.R.; Aslani, S.; Salmaninejad, A.; Javan, M.R.; Rezaei, N. PD-1/PD-L and autoimmunity: A growing relationship. Cell. Immunol. 2016, 310, 27–41.
- Nakazawa, A.; Dotan, I.; Brimnes, J.; Allez, M.; Shao, L.; Tsushima, F.; Azuma, M.; Mayer, L. The Expression and Function of Costimulatory Molecules B7h and B7-H1 on Colonic Epithelial Cells. Gastroenterology 2004, 126, 1347–1357.
- Faleiro, R.; Liu, J.; Karunarathne, D.; Edmundson, A.; Winterford, C.; Nguyen, T.H.; Simms, L.A.; Radford-Smith, G.; Wykes, M. Crohn’s disease is facilitated by a disturbance of programmed death-1 ligand 2 on blood dendritic cells. Clin. Transl. Immunol. 2019, 8.
- Cassol, C.A.; Owen, D.; Kendra, K.; Braga, J.R.; Frankel, W.L.; Arnold, C.A. Programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) expression in PD-1 inhibitor-associated colitis and its mimics. Histopathology 2020, 77, 240–249.
- Pinchuk, I.V.; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 Ligand Expression by Human Colonic Myofibroblasts/Fibroblasts Regulates CD4+ T-Cell Activity. Gastroenterology 2008, 135, 1228–1237.
- Beswick, E.J.; Grim, C.; Singh, A.; Aguirre, J.E.; Tafoya, M.; Qiu, S.; Rogler, G.; McKee, R.; Samedi, V.; Ma, T.Y.; et al. Expression of programmed death-ligand 1 by human colonic CD90+ stromal cells differs between ulcerative colitis and Crohn’s disease and determines their capacity to suppress Th1 cells. Front. Immunol. 2018, 9.
- Robertson, J.; Haas, C.T.; Pele, L.C.; Monie, T.P.; Charalambos, C.; Parkes, M.; Hewitt, R.E.; Powell, J.J. Intestinal APCs of the endogenous nanomineral pathway fail to express PD-L1 in Crohn’s disease. Sci. Rep. 2016, 6.
- Lina, T.T.; Alzahrani, S.; House, J.; Yamaoka, Y.; Sharpe, A.H.; Rampy, B.A.; Pinchuk, I.V.; Reyes, V.E. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 2015, 10, e121841.
- Beswick, E.J.; Pinchuk, I.V.; Das, S.; Powell, D.W.; Reyes, V.E. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect. Immun. 2007, 75, 4334–4341.
- Ponce de León, C.; Angel López-Casado, M.; Lorite, P.; Palomeque, T.; Isabel Torres, M. Dysregulation of the PD-1/PD-L1 pathway contributes to the pathogenesis of celiac disease. Cell. Mol. Immunol. 2019, 16, 777–779.
- Shan, T.; Chen, S.; Wu, T.; Yang, Y.; Li, S.; Chen, X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int. J. Clin. Exp. Pathol. 2019, 12, 1764–1769.
- Wyss, J.; Dislich, B.; Koelzer, V.H.; Galván, J.A.; Dawson, H.; Hädrich, M.; Inderbitzin, D.; Lugli, A.; Zlobec, I.; Berger, M.D. Stromal PD-1/PD-L1 Expression Predicts Outcome in Colon Cancer Patients. Clin. Colorectal Cancer 2019, 18, e20–e38.
- Zhao, Q.; Hu, F.; Xiao, Z.; Li, M.; Wu, X.; Zhao, Y.; Wu, Y.; Yin, J.; Lin, L.; Zhang, H.; et al. Comprehensive molecular profiling of the B7 family in gastrointestinal cancer. Cell Prolif. 2018, 51, e12468.
- Janakiram, M.; Chinai, J.M.; Fineberg, S.; Fiser, A.; Montagna, C.; Medavarapu, R.; Castano, E.; Jeon, H.; Ohaegbulam, K.C.; Zhao, R.; et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin. Cancer Res. 2015, 21, 2359–2366.
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298.
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112.
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated with PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40.
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020, 6, 1–21.
- Uhlig, H.H.; Powrie, F. Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu. Rev. Immunol. 2018, 36, 755–781.
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245.
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal cell PD-L1 inhibits CD8+ T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol. Res. 2018, 6, 1426–1441.
- Beswick, E.J.; Johnson, J.R.; Saada, J.I.; Humen, M.; House, J.; Dann, S.; Qiu, S.; Brasier, A.R.; Powell, D.W.; Reyes, V.E.; et al. TLR4 Activation Enhances the PD-L1–Mediated Tolerogenic Capacity of Colonic CD90+ Stromal Cells. J. Immunol. 2014, 193, 2218–2229.
- Aguirre, J.E.; Beswick, E.J.; Grim, C.; Uribe, G.; Tafoya, M.; Chacon Palma, G.; Samedi, V.; McKee, R.; Villeger, R.; Fofanov, Y.; et al. Matrix metalloproteinases cleave membrane-bound PD-L1 on CD90+ (myo-) fibroblasts in Crohn’s disease and regulate Th1/Th17 cell responses. Int. Immunol. 2020, 32, 57–68.
- Mezache, L.; Magro, C.; Hofmeister, C.; Pichiorri, F.; Sborov, D.; Nuovo, G.J. Modulation of PD-L1 and CD8 Activity in Idiopathic and Infectious Chronic Inflammatory Conditions. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 100–109.
- Maruya, M.; Kawamoto, S.; Kato, L.M.; Fagarasan, S. Impaired selection of IgA and intestinal dysbiosis associated with PD-1-deficiency. Gut Microbes 2013, 4, 165–171.
- Kawamoto, S.; Tran, T.H.; Maruya, M.; Suzuki, K.; Doi, Y.; Tsutsui, Y.; Kato, L.M.; Fagarasan, S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 2012, 336, 485–489.
- Rabe, H.; Nordström, I.; Andersson, K.; Lundell, A.C.; Rudin, A. Staphylococcus aureus convert neonatal conventional CD4+ T cells into FOXP3+ CD25+ CD127low T cells via the PD-1/PD-L1 axis. Immunology 2014, 141, 467–481.
- Scandiuzzi, L.; Ghosh, K.; Hofmeyer, K.A.; Abadi, Y.M.; Lázár-Molnár, E.; Lin, E.Y.; Liu, Q.; Jeon, H.; Almo, S.C.; Chen, L.; et al. Tissue-Expressed B7-H1 Critically Controls Intestinal Inflammation. Cell Rep. 2014, 6, 625–632.
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed cell death-1 receptor (Pd-1)-mediated regulation of innate lymphoid cells. Int. J. Mol. Sci. 2019, 20, 2836.
- Schwartz, C.; Khan, A.R.; Floudas, A.; Saunders, S.P.; Hams, E.; Rodewald, H.R.; McKenzie, A.N.J.; Fallon, P.G. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 2017, 214, 2507–2521.
- Reynoso, E.D.; Elpek, K.G.; Francisco, L.; Bronson, R.; Bellemare-Pelletier, A.; Sharpe, A.H.; Freeman, G.J.; Turley, S.J. Intestinal Tolerance Is Converted to Autoimmune Enteritis upon PD-1 Ligand Blockade. J. Immunol. 2009, 182, 2102–2112.
- Brown, J.A.; Dorfman, D.M.; Ma, F.-R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J. Immunol. 2003, 170, 1257–1266.
- Kryczek, I.; Wei, S.; Gong, W.; Shu, X.; Szeliga, W.; Vatan, L.; Chen, L.; Wang, G.; Zou, W. Cutting Edge: IFN-γ Enables APC to Promote Memory Th17 and Abate Th1 Cell Development. J. Immunol. 2008, 181, 5842–5846.
- Karakhanova, S.; Meisel, S.; Ring, S.; Mahnke, K.; Enk, A.H. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur. J. Immunol. 2009, 40, 254–266.
- Zhao, Q.; Xiao, X.; Wu, Y.; Wei, Y.; Zhu, L.-Y.; Zhou, J.; Kuang, D.-M. Interleukin-17-educated monocytes suppress cytotoxic T-cell function through B7-H1 in hepatocellular carcinoma patients. Eur. J. Immunol. 2011, 41, 2314–2322.
- Carol, M.; Lambrechts, A.; Van Gossum, A.; Libin, M.; Goldman, M.; Mascart-Lemone, F. Spontaneous secretion of interferon γ and interleukin 4 by human intraepithelial and lamina propria gut lymphocytes. Gut 1998, 42, 643–649.
- Troncone, E.; Marafini, I.; Stolfi, C.; Monteleone, G. Transforming growth factor-β1/Smad7 in intestinal immunity, inflammation, and cancer. Front. Immunol. 2018, 9, 1407.
- Garo, L.P.; Ajay, A.K.; Fujiwara, M.; Beynon, V.; Kuhn, C.; Gabriely, G.; Sadhukan, S.; Raheja, R.; Rubino, S.; Weiner, H.L.; et al. Smad7 Controls Immunoregulatory PDL2/1-PD1 Signaling in Intestinal Inflammation and Autoimmunity. Cell Rep. 2019, 28, 3353–3366.
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune Checkpoint Modulation in Colorectal Cancer: What’s New and What to Expect. J. Immunol. Res. 2015, 2015, 158038.
- Valentini, A.M.; Di Pinto, F.; Cariola, F.; Guerra, V.; Giannelli, G.; Caruso, M.L.; Pirrelli, M. PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironments. Oncotarget 2018, 9, 8584–8596.
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101.
- Siakavellas, S.I.; Bamias, G. Checkpoint inhibitor colitis: A new model of inflammatory bowel disease? Curr. Opin. Gastroenterol. 2018, 34, 377–383.
- Som, A.; Mandaliya, R.; Alsaadi, D.; Farshidpour, M.; Charabaty, A.; Malhotra, N.; Mattar, M.C. Immune checkpoint inhibitor-induced colitis: A comprehensive review. World J. Clin. Cases 2019, 7, 405–418.
- Bellaguarda, E.; Hanauer, S. Checkpoint Inhibitor-Induced Colitis. Am. J. Gastroenterol. 2020, 115, 202–210.
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168.
- Ibraheim, H.; Perucha, E.; Powell, N. Pathology of immune-mediated tissue lesions following treatment with immune checkpoint inhibitors. Rheumatology 2019, 58, vii17–vii28.
- Chen, J.H.; Pezhouh, M.K.; Lauwers, G.Y.; Masia, R. Histopathologic Features of Colitis Due to Immunotherapy with Anti-PD-1 Antibodies. Am. J. Surg. Pathol. 2017, 41, 643–654.
- Lee, S.A.; Wang, Y.; Liu, F.; Riordan, S.M.; Liu, L.; Zhang, L. Escherichia coli K12 upregulates PD-L1 expression in IFN-γ sensitized intestinal epithelial cells via the NF-κB pathway. Infect. Immun. 2020.
- Alpdundar Bulut, E.; Bayyurt Kocabas, B.; Yazar, V.; Aykut, G.; Guler, U.; Salih, B.; Surucu Yilmaz, N.; Ayanoglu, I.C.; Polat, M.M.; Akcali, K.C.; et al. Human Gut Commensal Membrane Vesicles Modulate Inflammation by Generating M2-like Macrophages and Myeloid-Derived Suppressor Cells. J. Immunol. 2020, 205, 2707–2718.
- Zhou, L.; Liu, D.; Xie, Y.; Yao, X.; Li, Y. Bifidobacterium infantis Induces Protective Colonic PD-L1 and Foxp3 Regulatory T Cells in an Acute Murine Experimental Model of Inflammatory Bowel Disease. Gut Liver 2019, 13, 430–439.
- Kennedy, L.C.; Bhatia, S.; Thompson, J.A.; Grivas, P. Preexisting autoimmune disease: Implications for immune checkpoint inhibitor therapy in solid tumors. J. Natl. Compr. Cancer Netw. 2019, 17, 750–757.
- Abu-Sbeih, H.; Faleck, D.M.; Ricciuti, B.; Mendelsohn, R.B.; Naqash, A.R.; Cohen, J.V.; Sellers, M.C.; Balaji, A.; Ben-Betzalel, G.; Hajir, I.; et al. Immune Checkpoint Inhibitor Therapy in Patients With Preexisting Inflammatory Bowel Disease. J. Clin. Oncol. 2020, 38, 576–583.
- Menzies, A.M.; Johnson, D.B.; Ramanujam, S.; Atkinson, V.G.; Wong, A.N.M.; Park, J.J.; McQuade, J.L.; Shoushtari, A.N.; Tsai, K.K.; Eroglu, Z.; et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann. Oncol. 2017, 28, 368–376.