The discovery of advanced materials is fundamentally transitioning from brute-force, database-dependent computational screening to targeted generative inverse design. High-Throughput Screening (HTS), powered by Density Functional Theory (DFT), provides a forward-mapping approach that remains constrained by the limits of known structural libraries. Conversely, deep generative models utilize artificial intelligence to navigate continuous chemical spaces via backward-mapping. This topic review explores the distinct mechanics of both paradigms, examining foundational chemical space representations alongside advanced deep learning architectures such as Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Diffusion models. Furthermore, it critically addresses the inherent computational bottlenecks of HTS and the ongoing challenge of material synthesizability in generative AI, charting the future trajectory of autonomous materials discovery.
- High-Throughput Screening
- Inverse Design
- Density Functional Theory
- Generative Adversarial Networks
- Variational Autoencoders
- Diffusion Models
- Latent Space
- Active Learning
- Synthesizability
- Crystal Graphs.
1. Introduction
The rational design and optimization of advanced functional materials dictate the pace of technological progression in energy storage, electrocatalysis, and nano-electronics.[1] Historically, materials discovery was dominated by stochastic, Edisonian methodologies reliant on empirical trial-and-error.[2] The maturation of first-principles quantum mechanical solvers, specifically Density Functional Theory (DFT), introduced the capability to predict electronic, thermodynamic, and structural properties in silico.[3]
Contemporary computational materials science is currently bifurcated into two distinct operational paradigms: High-Throughput Screening (HTS) and Deep Generative Inverse Design.[4] HTS operates as a forward-mapping deterministic filter, sequentially evaluating massive, predefined crystallographic databases. In contrast, Generative Inverse Design leverages advanced machine learning (ML) architectures to perform backward-mapping, directly outputting novel atomic coordinates optimized for targeted macroscopic properties.[5] This topic review establishes the mechanistic boundary between these frameworks, detailing their algorithmic foundations, thermodynamic scaling limitations, and implications for autonomous discovery.
2. Historical Milestones and Database Infrastructure
The transition toward automated high-throughput workflows was catalyzed in the early 2010s by the proliferation of massively parallel supercomputing architectures.[6] The establishment of the Materials Project in 2011 marked the inception of the HTS era, providing an open-access repository of DFT-calculated properties for tens of thousands of inorganic lattices.[7] Parallel databases, notably the Open Quantum Materials Database (OQMD)[8] and AFLOW[9], established the critical topological and thermodynamic baselines required for large-scale data mining.
By 2018, the materials science community recognized the fundamental limitations of discrete database mining: the calculated libraries represented only a fraction of the theoretical chemical space, which is estimated to exceed 1060 permutations for small molecules alone.[10][11] The application of Variational Autoencoders (VAEs) to chemical graphs demonstrated that discrete structures could be mapped into continuous, differentiable vector fields.[12] Between 2020 and the present, the field experienced a generative explosion. The adaptation of Generative Adversarial Networks (GANs) and Denoising Diffusion Probabilistic Models (DDPMs) extended inverse design capabilities from zero-dimensional molecular graphs to periodic, three-dimensional solid-state crystals.[13]
3. Methodological Mechanics of High-Throughput Screening
High-Throughput Screening operates on a deterministic forward-mapping principle, mathematically defined as:
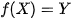
where a predefined atomic configuration (X) is subjected to a quantum mechanical Hamiltonian or surrogate model (f) to yield an observable property (Y).[14]
In practice, HTS functions as a multi-tier computational funnel.[15] A massive structural library—often generated via high-throughput elemental substitution within known stable prototypes (e.g., spinels or perovskites)—is subjected to sequential filters of increasing computational cost.[16] The primary tier strictly evaluates thermodynamic stability by computing the formation energy (ΔEf) relative to the elemental convex hull. Candidates exhibiting high energy above the hull (Ehull > 0) are rejected. Surviving candidates progress to intermediate tiers evaluating electronic band structures, while the final tier reserves highly expensive transition-state calculations (such as Nudged Elastic Band methods) for kinetic barrier assessment.
4. The Computational Bottleneck of Forward-Mapping
Despite its success in identifying functional electrolytes and photovoltaics, HTS exhibits rigid scaling limitations. The computational expenditure of HTS scales linearly (O(N)) with the library size. Because self-consistent field (SCF) calculations in standard DFT scale cubically (O(M3)) with the number of valence electrons, screening exhaustive compositional matrices requires prohibitive supercomputing resources.
The defining inefficiency of HTS is its resource allocation: the overwhelming majority of computational bandwidth is expended precisely computing the properties of non-viable materials destined for rejection. Furthermore, because HTS only interpolates within established crystallographic prototypes, it is fundamentally incapable of discovering non-intuitive, out-of-distribution structural archetypes.
5. The Paradigm Shift to Deep Generative Inverse Design
Deep Generative Inverse Design resolves the HTS bottleneck by defining an optimization-driven backward-mapping function:
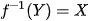
where the targeted functional property (Y) serves as the boundary condition, and the algorithm outputs the precise structural coordinates and stoichiometry (X) required to actualize it.
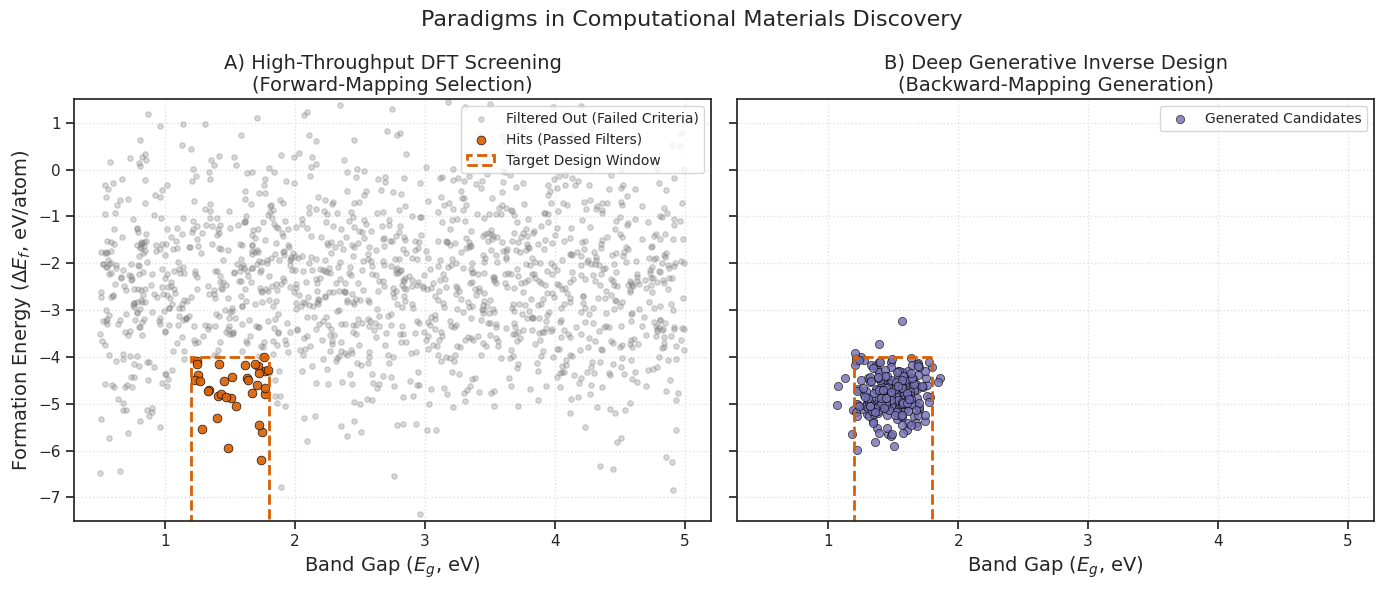
Figure 1. Paradigms in computational materials discovery. (A) The brute-force funnel of High-Throughput Screening (HTS), where computational resources are expended evaluating candidates that fail to meet property criteria. (B) Deep Generative Inverse Design, where targeted latent space sampling strictly populates the optimal property window, maximizing computational efficiency.
As illustrated in Figure 1, inverse design shifts the computational burden. Instead of expending resources verifying individual structures during the discovery phase, the computational cost is front-loaded into training the neural network.[17] Once the underlying physical distributions are learned, the model rapidly samples coordinates directly within the target parameter window.
6. Structural Representations and Latent Space Topology
Training neural networks on solid-state materials necessitates encoding periodic boundary conditions and rotational/translational invariances into machine-readable formats.
The field has standardized around Crystal Graph Convolutional Neural Networks (CGCNNs), which represent atoms as feature-rich nodes and interatomic distances as edges.[18] Generative models compress these high-dimensional crystal graphs into a continuous, low-dimensional "latent space". Within this mathematically smooth manifold, neighboring coordinates correspond to structurally analogous materials, allowing researchers to utilize gradient descent to computationally "walk" toward regions of maximized physical performance before decoding the coordinates back into a physical crystal.
7. Core Deep Learning Architectures
Three primary architectures dominate solid-state generative design:
-
Variational Autoencoders (VAEs): VAEs utilize an encoder to map structures to a Gaussian distribution in the latent space, optimized via the Evidence Lower Bound (ELBO), and a decoder to reconstruct the lattice. Joint property-prediction layers allow for condition-specific generation.
-
Generative Adversarial Networks (GANs): GANs operate via a minimax game. A Generator synthesizes hypothetical crystal structures from random noise, while a Discriminator evaluates adherence to implicit chemical rules (e.g., charge neutrality, Pauling’s rules) against real empirical data.
-
Denoising Diffusion Probabilistic Models (DDPMs): Operating via Langevin dynamics, diffusion models systematically corrupt atomic coordinates with Gaussian noise until thermodynamic equilibrium is reached. The network learns to reverse this Markov chain, denoising amorphous atomic clouds into highly ordered, symmetry-preserved, and thermodynamically optimized lattices.
8. Comparative Benchmark Analysis
The operational divergence between High-Throughput Screening (HTS) and Generative Inverse Design is not merely a difference in algorithmic efficiency; it represents a fundamental philosophical shift in how we mathematically represent and navigate the universe of synthesizable matter.
At its core, the division is a question of search space bounds, dimensional scaling, and structural novelty. HTS functions as a discrete grid search. It relies heavily on interpolating within the rigid boundaries of predefined stoichiometric families—for instance, systematically substituting transition metals within an AxByO3 perovskite framework. While this virtually guarantees that the resulting compounds will obey fundamental crystallographic rules, it heavily penalizes discovery. HTS is functionally blind to out-of-distribution structural archetypes; it cannot discover a completely novel lattice topology because that topology was never programmed into the initial screening database.
Conversely, Generative Inverse Design treats chemical space not as a discrete list of known compounds, but as a continuous, differentiable mathematical manifold. By mapping discrete crystal graphs into a continuous latent space, generative algorithms can extrapolate beyond known chemical boundaries. This allows the AI to traverse the "empty space" between established material classes, hypothesizing highly non-intuitive atomic configurations. Consequently, while HTS dominates in producing reliable, immediate variants of known materials, Inverse Design represents the only computationally viable pathway to discovering entirely new classes of advanced materials. The table below benchmarks the operational tradeoffs between these two paradigms.
| Performance Metric | High-Throughput Screening (HTS) | Deep Generative Inverse Design |
| Mathematical Mapping | Forward-mapping (f(X) = Y) | Backward-mapping (f-1 (Y) = X) |
| Search Space Bounds | Discrete, fixed, finite libraries | Continuous, fluid, theoretically infinite |
| Primary Computational Cost | Incurred during execution (DFT runs) | Incurred upfront during model training |
| Scaling Dependency | Scales linearly with database size (O(N)) | Scales with architecture and latent dimensions |
| Structural Novelty | Low; uncovers variants of known phases | High; discovers non-intuitive configurations |
| Synthesizability Rate | Extremely high; filters stable structures | Moderate to low; generates unstable anomalies |
| Data Requirements | Requires no initial training data | Requires massive, curated training datasets |
9. The Synthesizability Challenge and Active Learning
The critical bottleneck constraining Generative Inverse Design is the synthesizability gap. Unconstrained generative models frequently output mathematically optimal coordinates that reside in metastable or dynamically unstable regions of the potential energy surface. When rigorously evaluated via DFT, these structures exhibit high energy above the convex hull (Ehull), rendering physical laboratory synthesis impossible.
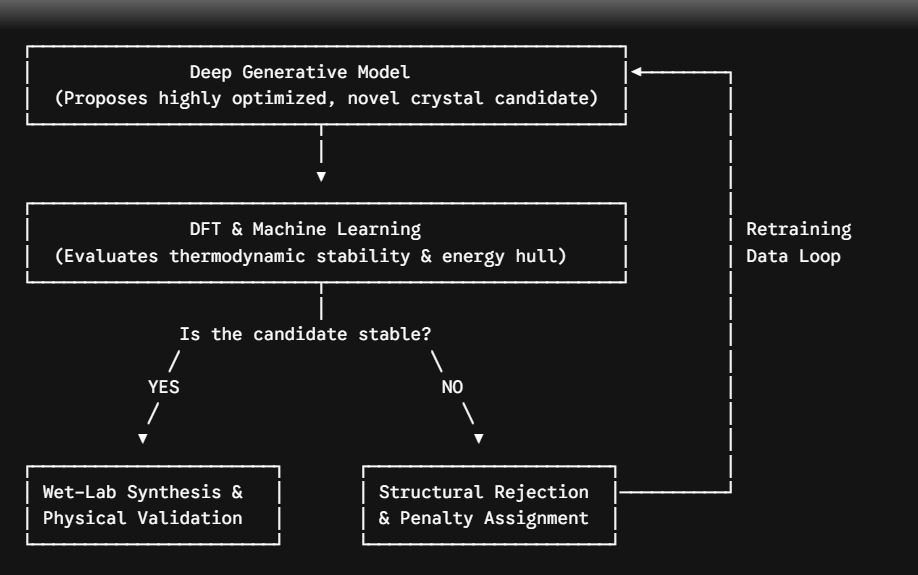
Figure 2. The Active Learning architecture for generative materials design. This closed-loop framework integrates generative proposal networks with rapid intermediate thermodynamic validation, utilizing failure data to iteratively improve the physical synthesizability of proposed candidates.
To enforce thermodynamic viability, modern discovery architectures deploy Active Learning loops (Figure 2). Generative proposals are passed through high-speed Machine Learning Interatomic Potentials (MLIPs) prior to full DFT relaxation. Candidates failing stability criteria are discarded, and the failure gradients are backpropagated into the generative model as structural penalties. This closed-loop iteration ensures the network iteratively converges on materials that are both functionally optimal and physically synthesizable.
10. Future Trajectories in Autonomous Discovery
At present, a distinct schism exists between industrial research and development (R&D) and the academic frontier. High-Throughput Screening remains the undisputed standard in industrial materials science. This dominance is driven by risk aversion and manufacturing realities: HTS exclusively outputs materials based on well-understood, thermodynamically stable crystallographic prototypes. Experimentalists and process engineers can readily conceptualize these outputs, adapt existing synthesis protocols, and scale them for commercial manufacturing. However, the academic sector has decisively pivoted toward Generative Inverse Design, recognizing that incremental structural screening is insufficient to overcome the compounding global demands for next-generation energy storage and green catalysis.
The immediate future of computational materials acceleration relies on the deployment of Multimodal Foundation Models. Rather than relying on isolated algorithms for property prediction and structural generation, the next generation of materials AI will synthesize Large Language Models (LLMs) directly with 3D crystal diffusion networks. These multimodal agents are being trained simultaneously on structural databases, computational thermodynamic parameters, and millions of pages of unstructured text from historical synthesis literature. This integration will allow researchers to input abstract, multi-constraint natural language prompts—such as, "Generate an earth-abundant, single-atom electrocatalyst stable in acidic media with a nitrogen reduction overpotential below 0.3 V"—and receive not just the mathematically optimized atomic coordinates, but a probabilistically generated, step-by-step laboratory synthesis recipe.
Ultimately, the true horizon of inverse design is the "Self-Driving Laboratory." The most persistent limitation of generative AI—the synthesizability gap—will be resolved by coupling digital generation directly with automated physical execution. In these closed-loop architectures, an AI proposes a novel material, which is subsequently validated by intermediate computational checks. The validated digital blueprint is then transmitted to an autonomous, robotic synthesis line capable of liquid-handling, high-temperature calcination, and automated X-ray diffraction (XRD) characterization. The robot physically attempts to synthesize the AI's hypothesized structure, characterizes the physical yield, and feeds the empirical success or failure data directly back into the generative model's loss function. By transforming theoretical candidate generation into physical, real-world deployment, these autonomous platforms will entirely remove human intuition as the rate-limiting step in materials discovery.
References
- Schmidt, J.; Marques, M.R.G.; Botti, S.; Marques, M.A.L. Recent advances and applications of machine learning in solid-state materials science. npj Comput. Mater.. 2019, 5, 83.
- Agrawal, A.; Choudhary, A. Perspective: Materials informatics and big data: Realization of the fourth paradigm of science.. APL Mater.. 2016, 4, 053208.
- Jain, A.; Shin, Y.; Persson, K.A. Computational challenges and strategies for the high-throughput discovery of materials.. Nat. Rev. Mater.. 2016, 1, 15004.
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design with generative models: Is a revolution underway?. Science. 2018, 361, 360–365.
- Zunger, A. Inverse design of materials. . Nat. Rev. Chem. . 2018, 2, 0121.
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater.. 2013, 12, 191–201.
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; Persson, K.A. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation.. APL Mater.. 2013, 1, 011002.
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM. 2013, 65, 1501–1509.
- Curtarolo, S.; Setyawan, W.; Wang, S.; Xue, J.; Yang, K.; Taylor, R.H.; Nelson, L.J.; Hart, G.L.W.; Sanvito, S.; Buongiorno-Nardelli, M.; Mingo, N.; Levy, O. AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci.. 2012, 58, 227–235.
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature. 2018, 559, 547–555.
- Reymond, J.L. The chemical space project. Acc. Chem. Res.. 2015, 48, 214–221.
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules.. ACS Cent. Sci. . 2018, 4, 268–276.
- Noh, J.; Kim, J.; Stein, H.S.; Sanchez-Lengeling, B.; Gregoire, J.M.; Aspuru-Guzik, A.; Jung, Y. Inverse design of solid-state materials via a continuous representation.. Matter. 2019, 1, 1370–1384.
- Takahashi, K.; Takahashi, L. Toward the Golden Age of Materials Informatics: Perspective and Opportunities. J. Phys. Chem. Lett. . 2023, 14, 4726–4733.
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalysts for hydrogen evolution. Nat. Mater. . 2006, 5, 909–913.
- Jain, A.; Voznyy, O.; Sargent, E.H. High-Throughput Screening of Lead-Free Perovskite-like Materials for Optoelectronic Applications. J. Phys. Chem. C. 2017, 121, 7183–7187.
- Xie, T.; Grossman, J.C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett.. 2018, 120, 145301.
- Huo, H.; Rupp, M. Unified representation of molecules and crystals for machine learning.. Machine Learning: Science and Technology. 2022, 3, 045011.