The downscaling of active sites to the atomic limit has revolutionized heterogeneous catalysis by maximizing atom utilization efficiency and creating highly uniform coordination centers. A comprehensive comparative analysis of Single-Atom Catalysts (SACs) and Dual-Atom Catalysts (DACs) across electrocatalytic, photocatalytic, and integrated photoelectrochemical applications reveals the distinct mechanistic advantages of multimetallic configurations. The coordination chemistry, electronic metal-support interactions (EMSI), and localized charge dynamics governing these systems dictate their catalytic efficiency. Critically, transitioning from isolated monometallic sites to synergetic homonuclear or heteronuclear diatomic centers breaks the classical adsorption scaling relations that restrict single-atom systems, defining the future trajectory of atomically dispersed catalyst design for complex multi-intermediate reactions.
- Single-Atom Catalysts
- Dual-Atom Catalysts
- Heterogeneous Catalysis
- Electrocatalysis
- Photocatalysis
- Scaling Relations
- Orbital Hybridization
- Density Functional Theory
- Charge Transfer
- X-ray Absorption Spectroscopy.
1. Introduction
The relentless pursuit of highly efficient, low-cost, and robust catalytic materials has driven the field of heterogeneous catalysis toward the atomic limit. In traditional nanocluster and bulk catalysts, only the surface atoms actively participate in the catalytic turnover, leaving the bulk interior electrochemically and photochemically dormant. The advent of atomically dispersed catalysts resolved this inefficiency, offering 100% atom utilization. Over the past decade, Single-Atom Catalysts (SACs) have emerged as a dominant frontier, exhibiting remarkable activity and selectivity due to their highly uniform coordination environments and distinct electronic structures.
However, as the focus of the scientific community shifts toward more complex, multi-electron, and multi-proton reactions—such as the Nitrogen Reduction Reaction (NRR), Carbon Dioxide Reduction Reaction (CO2RR), and the Oxygen Evolution Reaction (OER)—the intrinsic limitations of isolated monometallic sites have become apparent. To overcome these limitations, the paradigm is advancing toward Dual-Atom Catalysts (DACs). By anchoring two adjacent metal atoms (either homonuclear or heteronuclear) on a supporting matrix, DACs introduce synergistic orbital interactions and multi-site binding geometries that fundamentally alter reaction thermodynamics.
2. Structural Anatomy of Single-Atom Catalysts
The defining characteristic of a SAC is the absolute isolation of the metallic active site. In a SAC, a single transition metal atom (e.g., Pt, Fe, Co, Ni) is anchored to a solid support, completely lacking metal-metal coordination bonds. Instead, the atom is stabilized by strong electronic metal-support interactions (EMSI), typically forming coordination bonds with non-metal heteroatoms (such as nitrogen, oxygen, or sulfur) embedded within the supporting matrix.[1]
The most prevalent architecture is the M-N4 moiety (where M is the transition metal) embedded in a carbon framework, inspired by natural metalloporphyrins.[2] Beyond carbon matrices, highly structured frameworks such as Metal-Organic Frameworks (MOFs) and Covalent Organic Frameworks (COFs) provide ideal platforms for SACs. Their ordered porosity and tunable functional groups allow for the precise, defect-engineered spatial confinement of isolated metal atoms, preventing agglomeration under harsh electrocatalytic or photocatalytic operational conditions. Because all active sites are geometrically and electronically identical, SACs bridge the gap between homogeneous and heterogeneous catalysis, offering unprecedented selectivity.
3. The Limitations of the Single-Site Paradigm
Despite their maximized atom utilization, SACs are intrinsically constrained by classical thermodynamic limits known as scaling relations. In a complex catalytic cascade involving multiple adsorbed intermediates (e.g., *O, *OH, and *OOH in the OER), the binding energies of these intermediates are linearly correlated because they bind to the same singular metal atom through the same atomic orbital.[3]
Mathematically, if a SAC is structurally modified to bind *OH more strongly (to accelerate the initial activation step), it will simultaneously bind the subsequent *OOH intermediate too strongly, creating an insurmountable thermodynamic bottleneck during the product desorption phase. Because an isolated single atom possesses a singular d-band center, it lacks the geometrical and electronic flexibility to decouple the adsorption energies of varying intermediates. Consequently, a SAC often sits at a compromised position on the catalytic volcano plot, fundamentally incapable of minimizing the overpotential for complex multi-step transformations.
4. The Transition to Dual-Atom Catalysts
Dual-Atom Catalysts (DACs) resolve the structural rigidity of SACs by introducing a secondary, adjacent metallic site. DACs are broadly classified into two categories: homonuclear DACs (two identical metal atoms, e.g., Fe2-N6) and heteronuclear DACs (two different metal atoms, e.g., Fe/Co-N6).
The spatial arrangement of the diatomic pair is critical to its function. The two metal atoms can be directly bonded to one another (forming a localized metal-metal bond) or bridged by a shared heteroatom (such as a bridging oxygen or nitrogen).[4] This localized diatomic clustering fundamentally changes the active site from a single point of interaction to a highly synergistic coordination surface. The adjacency of the atoms allows them to operate in tandem, providing multiple spatial anchoring points for complex molecular reactants like N2 or CO2, which are difficult to activate on a single, isolated site. This fundamental structural evolution—from an isolated, electronically restricted single-metal center to a highly synergistic diatomic pair capable of cooperative binding—is visually summarized in Figure 1.
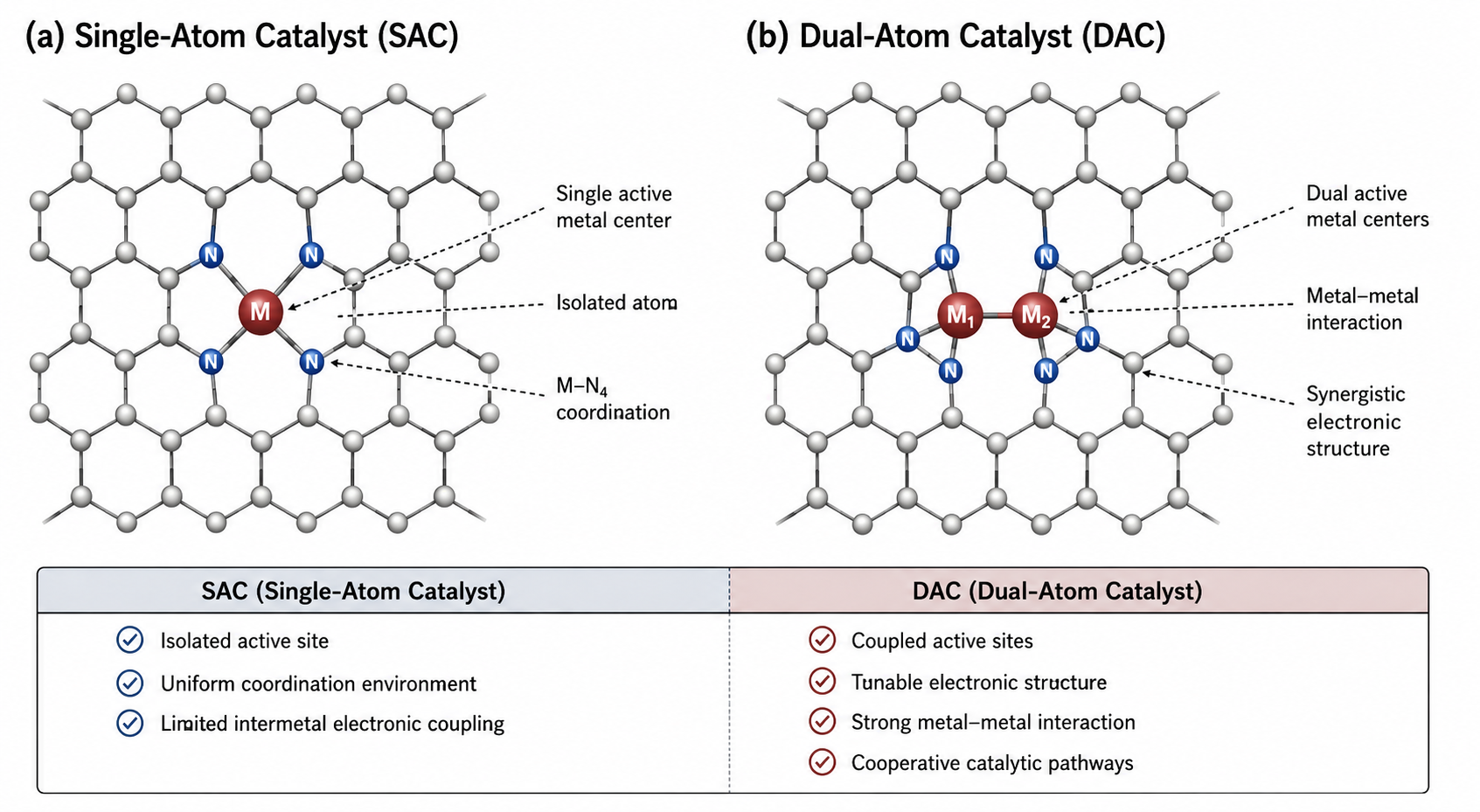
Figure 1. Structural comparison of atomically dispersed catalysts embedded within a nitrogen-doped carbon matrix. (a) A Single-Atom Catalyst (SAC) featuring an isolated transition metal center stabilized by a rigid M-N4 coordination environment with limited intermetal coupling. (b) A Dual-Atom Catalyst (DAC) featuring adjacent metal centers (M1-M2), introducing direct metal-metal interactions, tunable electronic structures, and cooperative active sites necessary for multi-step catalytic pathways.
5. Breaking Scaling Relations via Diatomic Synergy
The most profound mechanistic advantage of a DAC over a SAC is its thermodynamic ability to circumvent classical adsorption scaling relations. In electrocatalysis, the Sabatier principle dictates that the binding energy of an intermediate must be neither too strong nor too weak. However, on a monometallic single-atom site, the adsorption free energies (ΔG) of intermediates utilizing the same coordinating atom (e.g., *OH, *O, and *OOH in the oxygen evolution reaction) are rigidly coupled. For example, on a SAC, the relationship is fundamentally locked by the equation ΔG*OOH = ΔG*OH + 3.2 ± 0.2 eV. This universal 3.2 eV energy gap creates a theoretical minimum overpotential of roughly 0.4 V that a single atom cannot overcome, regardless of the supporting matrix.
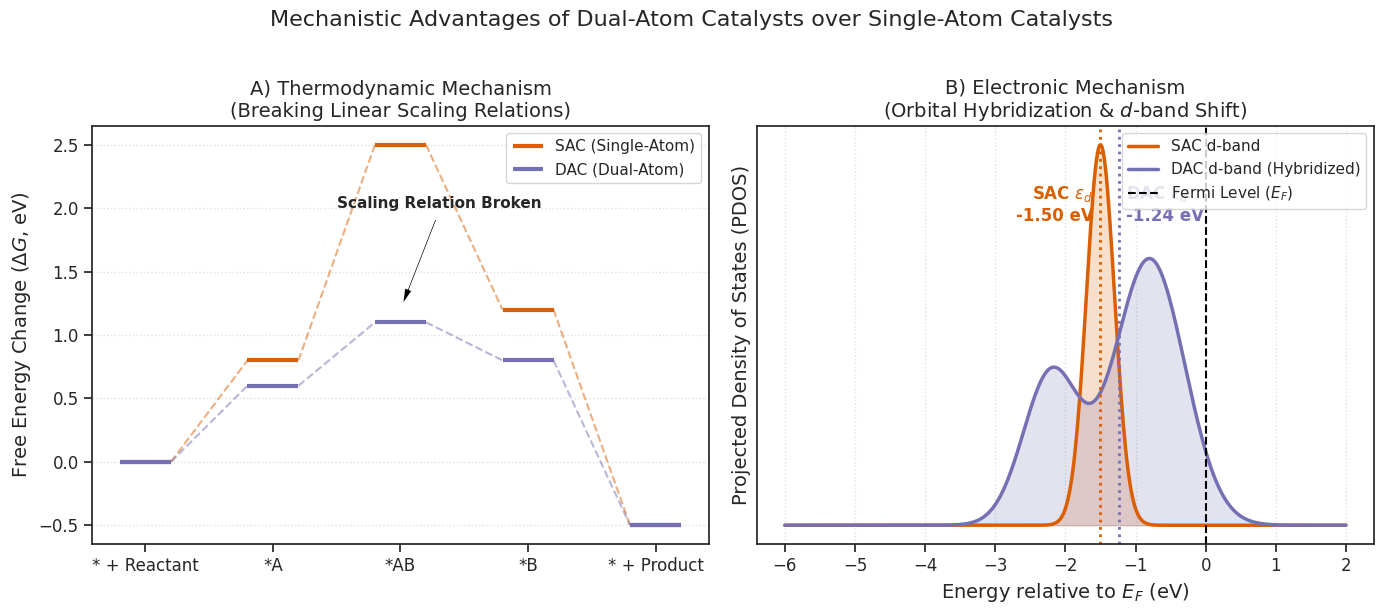
Figure 2. Mechanistic Advantages of Dual-Atom Catalysts over Single-Atom Catalysts. (A) Thermodynamic mechanism illustrating how a DAC breaks the linear scaling relation, selectively lowering the free energy barrier (ΔG) for a specific intermediate without penalizing the subsequent step. (B) Electronic mechanism showing the hybridization of d-orbitals in a DAC, resulting in the broadening of the density of states and a strategic shift of the d-band center (εd) relative to the Fermi level (EF).
As demonstrated in Figure 2A, a DAC provides dual-site binding modes that shatter this thermodynamic ceiling. In a heteronuclear DAC, the spatial decoupling allows the *OH intermediate to optimally bind to Metal A, while the bulkier *OOH intermediate bridges synergistically across Metal A and Metal B. This bi-dentate coordination stabilizes the *OOH state independently, narrowing the energy gap and lowering the overpotential.
Furthermore, as seen in Figure 2B, the physical proximity of the two atoms induces profound d-d orbital hybridization. Unlike a SAC, which features a highly localized, sharp d-band, the overlap of adjacent metal d-orbitals (such as the dz2 and dxz/dyz orbitals) in a DAC dramatically alters the projected density of states (PDOS). This hybridization broadens the d-band and shifts the d-band center (εd) relative to the Fermi level (EF). In heteronuclear DACs, the differing electronegativities of the two metals trigger asymmetric charge polarization, pushing localized charge depletion/accumulation zones that tune the binding strength of adsorbates precisely into the peak of the catalytic volcano plot.
6. Charge Dynamics in Photocatalysis
In the realm of photocatalysis and photoelectrocatalysis, the transition from SACs to DACs significantly enhances charge carrier dynamics. When a semiconductor support absorbs a photon, it generates electron-hole (e-/h+) pairs. In a SAC, the isolated metal atom acts as a highly efficient singular electron trap, funneling photogenerated electrons to the surface to drive reduction reactions while mitigating bulk recombination.[5]
However, DACs elevate this dynamic by establishing an internal electron-transfer bridge. In a heteronuclear pair, the distinct electronegativities of the two atoms create a localized internal electric field. Upon photoexcitation, electrons can rapidly migrate from the light-absorbing support to the primary metal, and subsequently to the secondary metal via intervalence charge transfer.[6][7] This unidirectional charge polarization drastically extends the lifetime of the excited state, ensuring a continuous supply of high-energy electrons for kinetically sluggish reactions, such as the photofixation of nitrogen.
7. Synthesis Strategies and Spatial Confinement
The fabrication of DACs is significantly more challenging than that of SACs, as the inherent high surface energy of diatomic pairs drives them toward agglomeration into larger nanoclusters. Top-down defect engineering and bottom-up molecular spatial confinement are the primary strategies utilized to lock DACs into place.
Standard synthesis often involves the high-temperature pyrolysis of metal-organic precursors. To achieve diatomic precision, researchers heavily rely on host-guest strategies, where bimetallic molecular precursors are entrapped within the highly defined pores of MOFs or COFs prior to carbonization.[8] Alternatively, atomic layer deposition (ALD) can be employed to sequentially anchor a second metal atom specifically onto a pre-existing single-atom site, leveraging the site-specific defect chemistry of the support.
8. Benchmark Electrocatalytic Applications and Mechanisms
The mechanistic superiority of DACs has resulted in breakthrough performances across several critical energy conversion pathways. The transition from single-point to dual-point coordination fundamentally alters the geometric pathways of elementary reaction steps.
8.1. Nitrogen Reduction Reaction (NRR)
The electrochemical synthesis of green ammonia is severely hindered by the ultra-stable N≡N triple bond (bond energy 941 kJ/mol) and the competing Hydrogen Evolution Reaction (HER). On a SAC, N2 can only adsorb via an end-on configuration (M-N≡N), which provides insufficient orbital overlap for efficient activation.[9] Homonuclear and heteronuclear DACs (such as Mo2 or Fe/Co centers) allow for a side-on bridging adsorption geometry (M-N-N-M). This bridging mode facilitates the simultaneous injection of electrons into the π* antibonding orbitals of N2 from both metal centers, drastically elongating and weakening the N-N bond and enabling an alternating hydrogenation pathway that is sterically impossible on a single isolated atom.
8.2. Carbon Dioxide Reduction Reaction (CO2RR)
The electrochemical reduction of CO2 to high-value C2+ products (such as ethylene or ethanol) requires the critical C-C coupling step. Because SACs possess only isolated active sites, they are functionally restricted to producing C1 products like carbon monoxide (CO) or formate (HCOOH); the physical distance between metal centers prevents two *CO intermediates from interacting. DACs directly solve this geographic limitation. The adjacent metal atoms naturally co-adsorb two *CO intermediates in close proximity, drastically lowering the kinetic barrier for asymmetric C-C dimerization and enabling the highly selective synthesis of multicarbon fuels.
8.3. Oxygen Electrocatalysis (ORR/OER)
In the Oxygen Reduction Reaction (ORR), DACs (particularly Fe/Co-N6 networks) routinely outperform commercial Pt/C benchmarks. Monometallic SACs often struggle with the sequential 2e- pathway, undesirably producing corrosive hydrogen peroxide (H2O2). DACs facilitate a dual-site O2 adsorption model where the O=O bond is stretched across both metal centers, promoting a highly efficient, direct 4e- reduction to water. In the reverse Oxygen Evolution Reaction (OER), the dual-site geometry allows for the direct coupling of two adjacent *O intermediates (O-O radical coupling mechanism), entirely bypassing the kinetically sluggish *OOH intermediate that limits SACs.
9. The Characterization Frontier and Future Directions
The definitive proof of diatomic adjacency remains the most critical hurdle in the field. Identifying a DAC requires advanced, state-of-the-art metrology. Spherical aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) is mandatory to visually resolve the sub-nanometer spacing between atom pairs. This must be coupled with Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy to quantitatively confirm the existence and coordination number of the specific metal-metal (M-M) bond pathway.
The future trajectory of atomically dispersed catalysts will increasingly rely on the integration of high-throughput computational screening, density functional theory, and machine learning to systematically pair metals from across the periodic table. By rationally matching the d-orbital symmetries of heteronuclear pairs, the rational design of DACs promises to finally overcome the fundamental thermodynamic ceilings of heterogeneous electro-photocatalysis.
References
- Wang, A., Li, J., Zhang, T. Heterogeneous single-atom catalysis.. Nat. Rev. Chem.. 2018, 2, 65-81.
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y., et al. Single-atom catalysis of CO oxidation using Pt1/FeOx.. Nat. Chem.. 2011, 3, 634-641.
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode.. J. Phys. Chem. B. 2004, 108, 17886-17892.
- Fu, J.; Dong, J.; Si, R.; et al. Synergistic Effects for Enhanced Catalysis in a Dual Single-Atom Catalyst. ACS Catalysis. 2021, 11, 1952-1961.
- Xia, Y.; Sayed, M.; Zhang, L.; Cheng, B.; Yu, J. Single-atom heterogeneous photocatalysts. . Chem Catalysis. 2021, 1, 1173-1214.
- Xue, Z.H.; Luan, D.; Zhang, H.; Lou, X.W.D. Single-atom catalysts for photocatalytic energy conversion.. Joule. 2022, 6, 92-133.
- Wu, J.; Zhong, H.; Huang, Z.F.; Zou, J.J.; Zhang, X.; Zhang, Y.C.; Pan, L. Research progress of dual-atom site catalysts for photocatalysis. . Nanoscale. 2024, 16, 9169-9185.
- Su, J.; Ge, R.; Dong, Y.; Hao, F.; Chen, L. Recent progress in single-atom electrocatalysts: concept, synthesis, and applications in clean energy conversion.. J. Mater. Chem. A. 2018, 6, 14025-14042.
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-atom catalysts: synthetic strategies and electrochemical applications.. Joule. 2018, 2, 1242-1264.