Definition (Draft for you)
Paracoccidioidomycosis is a neglected disease that causes economic and social impacts, mainly affecting people of certain social segments, such as rural workers. The limitations of antifungals, such as toxicity, drug interactions, restricted routes of administration, and the reduced bioavailability in target tissues, have become evident in clinical settings. These factors, added to the fact that Paracoccidioidomycosis (PCM) therapy is a long process, lasting from months to years, emphasize the need for the research and development of new molecules.
- Antifungal Drugs against Paracoccidioidomycosis
Thanks so much for your check. We sincerely hope you may create this entry, since you are the expert in this academic research. You can click the “submit” button to upload it after revision. We will help you layout after you submit it. Moreover, we will link your article at the entry, so more scholars and students can look through and cite it.
1. Introduction
The infection caused by fungi of the genus Paracoccidioides was first described by Adolpho Lutz in 1908. In 1971, during the meeting of several mycologists from Latin America in Medellín-Colombia, the term Paracoccidioidomycosis (PCM) was made official to designate the systemic granulomatous infection caused by the thermodimorphic fungi of the genus Paracoccidioides [1,2][1][2]. Several aspects of the infection remained unknown for a long period, including the taxonomic classification of the pathogen and a more appropriate therapeutic approach [3][3]. Nowadays, however, the importance of this disease in Latin America is recognized, with it being one of the main causes of deaths due to fungi infections [4][4].
The eradication of the fungus in the tissues is slow and treatment might last from months to years of antifungal administration. Paracoccidioides spp. are sensitive to various systemic antifungals, but the therapeutic options are limited to antifungals that act on two main targets, plasma membrane and folic acid synthesis [5][5]. Several new compounds with antifungal properties have been proposed against PCM over the last decade. Thus, this review focus on studies that have identified alternative compounds to the current treatment of PCM, as well as the strategies used for the development of new antifungal drugs.
2. Treatment of Paracoccidioidomycosis: An Overview
Figure 1 shows the historical context that defines the treatment of PCM currently. The first alternative treatment emerged in 1940, 32 years after the first reports of the disease by Lutz, using sulfapyridine, which is a sulfonamide derivative [6][6]. Sulfonamides are recognized for their broad spectrum of antibiotic activity, interrupting the growth of microorganisms by the competitive inhibition of the aminobenzoic acid (PABA) to the enzyme dihydropteroate synthase in the folate synthesis pathway. The latter is an essential component in the synthesis of nucleic acids and proteins [7,8][7][8]. Sulfa derivatives were also used in the treatment of PCM according to the severity of the disease, including compounds of low, moderate, and high excretion by the body [5][5].
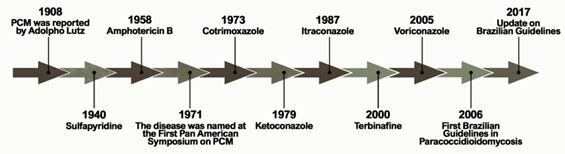
Figure 1.
Important events related to the treatment and establishment of a consensus on Paracoccidioidomycosis.
Treatment with amphotericin B (AmB) was introduced by Lacaz and Sampaio[6] [6] in 1958. This drug was established as an antifungal agent with a wide spectrum of action, acting mainly through the formation of complexes with ergosterol. These are molecules present in the membranes of fungal cells and responsible for the formation of transmembrane channels that allow the extravasation of cytoplasmic components [9][9]. It is indicated for severe and disseminated forms. Despite its effectiveness, AmB is also able to bind, to a lesser extent, to the cholesterol present in mammalian cells, producing several toxic effects on the host during the course of treatment, including acute symptoms such as nausea, vomiting, fever, hyper and hypotension, and hypoxia, in addition to chronic nephrotoxicity [10,11][10][11]. New formulations of AmB were developed for incorporation into liposomes, resulting in a better tissue distribution and less toxicity [5][5].
Another treatment option for PCM emerged with the introduction of cotrimoxazole (CMX) in 1973. This drug is a synergistic association between sulfamethoxazole (a sulfonamide derivative) and trimethoprim, and is used in patients with mild to moderate forms of PCM and neuroparacoccidioidomycosis [12–14][12][13][14]. CMX acts by inhibiting the enzymes involved in the synthesis of tetrahydrofolic acid, leading to the depletion of intracellular folate, which is essential for the growth of the pathogenic organism [15][15].
The development of azole derivatives certainly supported the expansion of the arsenal of antifungals against PCM. These compounds are the most common agents used in the treatment and prevention of a wide spectrum of mycoses, preventing the biosynthesis of ergosterol by inhibiting the CYP450-dependent enzyme, lanosterol 14-α-demethylase [16,17]. Two years after its publication in 1979, ketoconazole was introduced as an antifungal for the treatment of PCM [18–20]. Despite the success of this drug in controlling mild and moderate forms of the disease, its use is no longer recommended due to hepatotoxicity and adrenal insufficiency [21]. Thus, ketoconazole was eventually replaced by the first generation of triazoles, especially after the introduction of itraconazole (ITZ) in 1987 [22][16].
Some other alternative treatments have been proposed over the past two decades. A case report published in 2000 showed a patient who did not respond to the initial treatment with CMX but achieved clinical, mycological, and radiological cures two years after the end of treatment with terbinafine [23][17]. Terbinafine is an antifungal of the allylamine class that has a similar mechanism of action to azole agents, blocking the ergosterol biosynthesis pathway by inhibiting the squalene enzyme epoxidase [24][18]. A representative of the second-generation triazoles known as voriconazole has a similar efficacy to ITZ, and is useful in the treatment of neuro-PCM due to the greater penetration of this drug into the central nervous system compared to ITZ[19] [25].
The publication of the consensus on PCM in 2006 allowed the creation of guidelines to formalize the PCM clinical treatment. Shikanai-Yasuda recommends the use of ITZ as the drug of choice for the treatment of mild and moderate forms of PCM, followed by CMX and AmB, according to the severity of the disease. The consensus also points to the possibility of using voriconazole, posaconazole, and isavuconazole to replace itraconazole, paying attention to costs, clinical evidence, and drug interactions [14][14].
Even after one hundred years of investigation into the disease, the therapeutic approaches face several issues. The main problem to overcome concerns the long period of treatment required by the currently available antifungals, which occasionally results in patients giving up therapy [26][20]. Another issue is the possibility of Paracoccidioides developing resistance against these antifungals. In Paracoccidioides spp., genes that were homologous to the cerebellar degeneration-related protein (CDR1, CDR2) and multi-drug resistance (MDR1) of Candida albicans, the pleiotropic drug resistance (PDR5) of Saccharomyces cerevisiae, and the ABC transporter genes of Aspergillus spp. were observed; all of them were related to resistance against azoles [27][21]. Thus, as the current treatment against PCM is mainly based on azole derivatives, these genes may play a similar role in Paracoccidioides spp., with the possibility of developing resistant isolates. In fact, the in vitro resistance of Paracoccidioides spp. was evidenced against azole derivatives [28][22]. Cermeño et al. carried out a sensitivity test of several species of Paracoccidioides spp. and observed the resistance against caspofungin (94.7%), followed by 5-flucytosine (52.6%) and AmB (47.4%) [29][23].
In addition to antifungal therapy, the treatment of possible sequelae of PCM such as pulmonary fibrosis and the prevention of opportunistic diseases should also be considered. An additional therapy proposal for PCM aimed at reducing pulmonary fibrosis is the combination of itraconazole-pentoxifylline. However, although the combination has shown promising results in mice, there are still no reports on testing in humans.
3. New Anti-Paracoccidioides Compounds: Do We Already Have Any Ideal Antifungals?
The ideal antifungal agent should have a broad activity; be selective for fungal targets; and have reduced adverse effects, limited interactions, many administration routes, and low resistance. Thus, proteins belonging to the metabolic pathways involved in amino acid metabolism, cell wall, ergosterol biosynthesis, response to oxidative stress, and alternative sources of carbon are targets for antifungals and explored in the process of identifying new selective compounds. Such parameters are essential for the survival of the pathogen and additionally those targets should be absent in humans [30]. Over the years, several techniques and approaches helped to search for promising antifungals (Figure 2).
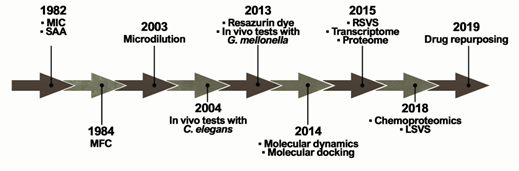
Figure 2. Methodologies used in the search and evaluation of anti-paracoccidioidomycosis compounds. MIC (Minimal Inhibitory Concentration), SAA (Synergistic Activity Assay), MFC (Minimal Fungicidal Activity), RSVS (Receptor shape-based Virtual Screening), LSVS (Ligand shape-based Virtual Screening).
The ability of a compound to inhibit fungal growth is known as Minimum Inhibitory Concentration (MIC). MIC was first used in a PCM study in 1982 to assess the in vitro effects of CMX against P. brasiliensis clinical isolates [31][24]. However, one of the major problems is the lack of standardization in these tests, which is directly related to the variety of MIC protocols found in the literature. Experimentally, tests that identify MIC values have been performed for several years through the macrodilution technique or by counting colony-forming units. This technique was improved by converting these assays to microdilution using 96-well microplates in 2003 [32][25]. In addition, the proposition of using resazurin dye as a marker of metabolic activity provided even greater practicality for those assays in 2013 [33][26]. Studies reported that certain variables, such as culture medium and incubation time, have a major impact on the results [34][27]. Although there is a trend towards the use of the RPMI-1640 culture medium, as recommended by the Clinical and Laboratory Standards Institute (CLSI) [35][28], the incubation time still remains undefined, ranging from 3 to 15 days of incubation [36,37][29][30].
The search for new antifungal candidates has been driven by synthetic, semi-synthetic, plant. and microorganism compounds. In this review, we describe some of the compounds tested against Paracoccidioides spp., and additional compounds are listed in Tables S1 and S2.
Potential compounds act as growth inhibitors against Paracoccidioides spp. in low concentrations (Table S1), such as alkyl gallates (0.004–16 µg/mL), an N-Glycosylation inhibitor [38,39][31][32]; the thiosemicarbazone lapachol, which acts on the plasma membrane (0.01–0.1 µM) [40][33]; azasterol analogs (0.5–10 µM)[34] [41] and hydrazone derivatives (0.1–5 µM) [42][35], as inhibitors of ergosterol biosynthesis. These compounds are less potent when compared to azole derivatives, such as itraconazole with a 0.003–0.05 µM MIC [33]; luliconazole, a topical antifungal repositioned against Paracoccidioides spp. through in vitro assays and with an MIC ranging from 0.0005 to 0.0007 µM [43][36]; and butaconazole, which is used for the local treatment of vulvovaginal candidiasis and also repositioned against Paracoccidioides spp. with an MIC of 0.001–0.002 µM. Both compounds are inhibitors of the ergosterol biosynthesis, indicating that this molecular target is very promising. Antimicrobial peptides have also been proposed as potent inhibitors of Paracoccidioides spp., such as MK58911, a peptide analogue of mastoparan, which presented an MIC of 7.8 μg/mL against P. brasiliensis and 15.6 μg/mL against P. lutzii [44][37]. Lactoferrin-derived peptides presented MIC values between 0.63 and 1.25 μg/mL [45][38].
The medicinal potential of plants as antifungals comes from their extracts, essential oils, and chemical constituents [46][39]. Several studies have reported compounds with antifungal properties against PCM, such as argentilactone derived from Hyptis ovalifolia [47][40], oenothein B derived from Eugenia uniflora [48,49][41][42], fatty acid methyl esters and compounds derived from Annona cornifolia essential oils [50][43], hydroalcoholic extracts from the species Piper regnellii and Baccharis dracunculifolia [51[44][45],52], and curcumin from Curcuma longa [53][46]. Another natural compound is ajoene, derived from Allium sativum, which exerts inhibitory effects against morphological transition and in yeast cells, with possible involvement in the sulfhydryl metabolism of P. brasiliensis [54][47].
Exploring the universe of natural source compounds, microorganism-derived compounds were tested against Paracoccidioides spp. (Table S2). The extracts and cytochalasin E isolated from Aspergillus felis presented an MIC of 31.2 µg/mL (3.6 µM) [55][48]. The extract of the endophytic fungus Fusarium sp. containing T2-toxin was able to inhibit clinical strains of P. brasiliensis with an MIC ranging between 75 and 640 nM and the extract containing 8-n-butyrylneosolaniol and 8-isobutyrylsolaniol had MIC values from 160 to 640 nM [56][49]. Altenusin, isolated from the endophytic fungus Alternaria sp., exhibited an MIC between 1.9 and 31.2 μg/mL [57][50]. Another important compound is farnesol, a C. albicans quorum sensing molecule, which acts as a potent antifungal inhibiting the growth and dimorphism in μM concentrations [58][51].
References
- Lutz, A. Uma micose pseudococcídica localizada na boca e observada no Brasil. Contribuição ao conhecimento das hifoblastomicoses americanas. In Adolpho Lutz—Dermatologia e Micologia; Editora FIOCRUZ: Rio de Janeiro, Brazil, 2004; Volume 1, p. 620; ISBN 85-7541-042-3.
- Marques, S.A. Paracoccidioidomicose: Centenário do primeiro relato de caso. An. Bras. Dermatol. 2008, 83, 271–273, doi:10.1590/S0365-05962008000300014.
- Lacaz, C.S. Historical Evolution of the Knowledge on Paracoccidioidomycosis and its Etiologic Agent, Paracoccidioides. In Paracoccidioidomycosis; Franco, M., da S. Lacaz, C., Restrepo-Moreno, A., del Negro, G., Eds.; CRC Press: Boca Ratón, FL, USA, 1994; pp. 1–22.
- Rodrigues, M.L.; Albuquerque, P.C. Searching for a change: The need for increased support for public health and research on fungal diseases. PLoS Negl. Trop. Dis. 2018, 12, e0006479, doi:10.1371/journal.pntd.0006479.
- Shikanai-Yasuda, M.A. Paracoccidioidomycosis treatment. Ver. Inst. Med. Trop. São Paulo 2015, 57, 31–37, doi:10.1590/S0036-46652015000700007.
- Mendes, R.P.; Cavalcante, R.S.; Marques, S.A.; Marques, M.E.A.; Venturini, J.; Sylvestre, T.F.; Paniago, A.M.M.; Pereira, A.C.; da Silva, J. de F.; Fabro, A.T.; et al. Paracoccidioidomycosis: Current Perspectives from Brazil. Open Microbiol. J. 2017, 11, 224–282, doi:10.2174/1874285801711010224.
- Saleem, H.; Maryam, A.; Bokhari, S.A.; Ashiq, A.; Rauf, S.A.; Khalid, R.R.; Qureshi, F.A.; Siddiqi, A.R. Design, synthesis, characterization and computational docking studies of novel sulfonamide derivatives. EXCLI J. 2018, 17, 169–180, doi:10.17179/excli2017-886.
- Chen, J.; Xie, S. Overview of sulfonamide biodegradation and the relevant pathways and microorganisms. Sci. Total Environ. 2018, 640–641, 1465–1477, doi:10.1016/j.scitotenv.2018.06.016.
- Adler-Moore, J.P.; Gangneux, J.-P.; Pappas, P.G. Comparison between liposomal formulations of amphotericin B. Med. Mycol. 2016, 54, 223–231, doi:10.1093/mmy/myv111.
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227, doi:10.1016/j.riam.2009.06.003.
- Kyriakidis, I.; Tragiannidis, A.; Munchen, S.; Groll, A.H. Clinical hepatotoxicity associated with antifungal agents. Expert Opin. Drug Safety 2017, 16, 149–165, doi:10.1080/14740338.2017.1270264.
- Barbosa, W.; de P. Vasconcelos, W.M. Ação da sulfametoxazol associada ao trimetoprim na terapêutica da blastomicose sul-americana. J. Trop. Pathol. 1973, 329-339, doi:10.5216/rpt.v2i3.22726.
- Capasso, C.; Supuran, C.T. Dihydropteroate synthase (sulfonamides) and dihydrofolate reductase inhibitors. In Bacterial Resistance to Antibiotics—From Molecules to Man; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2019; pp. 163–172; ISBN 978-1-119-59352-2.
- Shikanai-Yasuda, M.A.; Mendes, R.P.; Colombo, A.L.; de Queiroz-Telles, F.; Kono, A.S.G.; Paniago, A.M.M.; Nathan, A.; do Valle, A.C.F.; Bagagli, E.; Benard, G.; et al. Brazilian guidelines for the clinical management of paracoccidioidomycosis. Rev. Soc. Bras. Med. Trop.2017, 50, 715–740, doi:10.1590/0037-8682-0230-2017.
- Smilack, J.D. Trimethoprim-sulfamethoxazole. Mayo Clin. Proc. 1999, 74, 730–734, doi:10.4065/74.7.730.
- Negroni, R.; Palmieri, O.; Koren, F.; Tiraboschi, I.N.; Galimberti, R.L. Oral treatment of paracoccidioidomycosis and histoplasmosis with itraconazole in humans. Rev. Infect. Dis. 1987, 9 (Suppl. 1), S47–S50, doi:10.1093/clinids/9.supplement_1.s47.
- Ollague, J.M.; Zurita, A.M.D.; Calero, G. Paracoccidioidomycosis (South American blastomycosis) successfully treated with terbinafine: First case report. Br. J. Dermatol. 2000, 143, 188–191, doi:10.1046/j.1365-2133.2000.03614.x.
- Visbal, G.; San-Blas, G.; Murgich, J.; Franco, H. Paracoccidioides brasiliensis, paracoccidioidomycosis, and antifungal antibiotics. Curr. Drug Targets Infect. Disord. 2005, 5, 211–226, doi:10.2174/1568005054880118.
- Telles, F.Q.; Goldani, L.Z.; Schlamm, H.T.; Goodrich, J.M.; Ingroff, A.E.; Yasuda, M.A.S. An Open-Label comparative pilot study of oral voriconazole and itraconazole for long-term treatment of paracoccidioidomycosis. Clin. Infect. Dis. 2007, 45, 1462–1469, doi:10.1086/522973.
- Andrade, U.V.; Oliveira, S.M.; Chang, M.R.; Pereira, E.F.; Marques, A.P.; de Carvalho, L.R.; Mendes, R.P.; Paniago, A.M.M.; Andrade, U.V.; Oliveira, S.M.; et al. Treatment compliance of patients with paracoccidioidomycosis in Central-West Brazil. J. Bras. Pneumol. 2019, 45, doi:10.1590/1806-3713/e20180167.
- Costa, C.; Albuquerque, F.C.; Andrade, R.V.; de Oliveira, G.C.; de Almeida, M.F.; Brigido, M; Maranhão, A.Q. Transporters in the Paracoccidioides brasiliensis transcriptome: Insights on drug resistance. Genet. Mol. Res. 2005, 4, 390–408.
- Hahn, R.C.; Morato Conceição, Y.T.; Santos, N.L.; Ferreira, J.F.; Hamdan, J.S. Disseminated paracoccidioidomycosis: Correlation between clinical and in vitro resistance to ketoconazole and trimethoprim sulphamethoxazole. Mycoses 2003, 46, 342–347. 10.1046/j.1439-0507.2003.00901.x.
- Cermehol, J.R.; Alvarado, P.; Mendoza, M.; Herndndez, I.; Cuestal, D. In vitro susceptibility of isolates of Paracoccidioides spp complex to systemic antifungals using the microdilution method. Investig. Clin. 2015, 56, 243–253.
- Stevens, D.A.; Vo, P.T. Synergistic interaction of trimethoprim and sulfamethoxazole on Paracoccidioides brasiliensis. Antim. Agents Chemother. 1982, 21, 852–854, doi:10.1128/AAC.21.5.852.
- Nakai, T.; Uno, J.; Ikeda, F.; Tawara, S.; Nishimura, K.; Miyaji, M. In vitro antifungal activity of micafungin (fk463) against dimorphic fungi: Comparison of yeast-like and mycelial forms. AAC 2003, 47, 1376–1381, doi:10.1128/AAC.47.4.1376-1381.2003.
- de Paula e Silva, A.C.A.; Oliveira, H.C.; Silva, J.F.; Sangalli-Leite, F.; Scorzoni, L.; Fusco-Almeida, A.M.; Mendes-Giannini, M.J.S. Microplate alamarblue assay for Paracoccidioides susceptibility testing. J. Clin. Microbiol. 2013, 51, 1250–1252, doi:10.1128/JCM.02914-12.
- Cruz, R.C.; Werneck, S.M.C.; Oliveira, C.S.; Santos, P.C.; Soares, B.M.; Santos, D.A.; Cisalpino, P.S. Influence of different media, incubation times, and temperatures for determining the mics of seven antifungal agents against Paracoccidioides brasiliensis by microdilution. J. Clin. Microbiol. 2013, 51, 436–443, doi:10.1128/JCM.02231-12.
- CLSI. CLSI-Clinical and Laboratory Standards Institute Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard. Document M27; CLSI: Wayne, PA, USA, 2008.
- de Sá, N.P.; Cisalpino, P.S.; Bertollo, C.M.; Santos, P.C.; Rosa, C.A.; de Souza, D.G.; Barbeira, P.J.S.; Alves, T.M.; Zani, C.L.; Johann, S. Thiosemicarbazone of lapachol acts on cell membrane in Paracoccidioides brasiliensis. Med. Mycol. 2019, 57, 332–339, doi:10.1093/mmy/myy045.
- do Carmo Silva, L.; Miranda, M.A.C.M.; de Freitas, J.V.; Ferreira, S.F.A.; de Oliveira Lima, E.C.; de Oliveira, C.M.A.; Kato, L.; Terezan, A.P.; Rodriguez, A.F.R.; Faria, F.S.E.D.V.; et al. Antifungal activity of Copaíba resin oil in solution and nanoemulsion against Paracoccidioides spp. Braz. J. Microbiol. 2020, 51, 125–134, doi:10.1007/s42770-019-00201-3.
- de Paula e Silva, A.C.A.; Costa-Orlandi, C.B.; Gullo, F.P.; Sangalli-Leite, F.; de Oliveira, H.C.; Silva, J.; Scorzoni, L.; Pitangui, N.; Rossi, S.A.; Benaducci, T.; et al. Antifungal activity of decyl gallate against several species of pathogenic fungi. Evid. Based Complem. Altern. Med. 2014, 2014, 1–8, doi:10.1155/2014/506273.
- de Paula e Silva, A.C.A.; de Oliveira, H.C.; Scorzoni, L.; Marcos, C.M.; dos Santos, C.T.; Fusco-Almeida, A.M.; Salina, A.C.G.; Medeiros, A.I.; Almeida, F.; Li, S.C.; et al. Decyl gallate as a possible inhibitor of n-glycosylation process in Paracoccidioides lutzi. Antimicrob. Agents Chemother. 2019, 63, e01909-18, doi:10.1128/AAC.01909-18.
- Souza, M.A.; Johann, S.; Lima, L.A.R.; Campos, F.F.; Mendes, I.C.; Beraldo, H.; de Souza-Fagundes, E.M.; Cisalpino, P.S.; Rosa, C.A.; Alves, T.M.; et al. The antimicrobial activity of lapachol and its thiosemicarbazone and semicarbazone derivatives. Memórias Inst. Oswaldo Cruz 2013, 108, 342–351, doi:10.1590/S0074-02762013000300013.
- Visbal, G.; Alvarez, A.; Moreno, B.; San-Blas, G. S-Adenosyl-l-Methionine Inhibitors Δ24-Sterol Methyltransferase and Δ24(28)-sterol methylreductase as possible agents against Paracoccidioides brasiliensis. AAC 2003, 47, 2966–2970, doi:10.1128/AAC.47.9.2966-2970.2003.
- Visbal, G.; San-Blas, G.; Maldonado, A.; Álvarez-Aular, Á.; Capparelli, M.V.; Murgich, J. Synthesis, in vitro antifungal activity and mechanism of action of four sterol hydrazone analogues against the dimorphic fungus Paracoccidioides brasiliensis. Steroids 2011, 76, 1069–1081, doi:10.1016/j.steroids.2011.04.012.
- de Oliveira, A.A.; Neves, B.J.; Silva, L.; Soares, C.M.; Andrade, C.H.; Pereira, M. Drug repurposing for paracoccidioidomycosis through a computational chemogenomics framework. Front. Microbiol. 2019, 10, 1301, doi:10.3389/fmicb.2019.01301.
- Singulani, J.L.; Galeane, M.C.; Ramos, M.D.; Gomes, P.C.; dos Santos, C.T.; de Souza, B.M.; Palma, M.S.; Fusco Almeida, A.M.; Mendes Giannini, M.J.S. Antifungal Activity, toxicity, and membranolytic action of a mastoparan analog peptide. Front. Cell. Infect. Microbiol. 2019, 9, 419, doi:10.3389/fcimb.2019.00419.
- Fernandes, K.E.; Carter, D.A. The antifungal activity of lactoferrin and its derived peptides: Mechanisms of action and synergy with drugs against fungal pathogens. Front. Microbiol. 2017, 8, doi:10.3389/fmicb.2017.00002.
- Sortino, M.; Derita, M.; Svetaz, L.; Raimondi, M.; Di Liberto, M.; Petenatti, E.; Gupta, M.; Zacchino, S. The role of natural products in discovery of new anti-infective agents with emphasis on antifungal compounds. Plant Bioact. Drug Discov. Princ. Pract. Persp. 2012, 4, 205–239. https://doi.org/10.1002/9781118260005.ch6.
- Silva, L.C.; Tauhata, S.B.F.; Baeza, L.C.; de Oliveira, C.M.A.; Kato, L.; Borges, C.L.; de Almeida Soares, C.M.; Pereira, M. Argentilactone molecular targets in Paracoccidioides brasiliensis identified by chemoproteomics. Antimicrob. Agents Chemother. 2018, 62, doi:10.1128/AAC.00737-18.
- Zambuzzi-Carvalho, P.F.; Tomazett, P.K.; Santos, S.C.; Ferri, P.H.; Borges, C.L.; Martins, W.S.; de Almeida Soares, C.M.; Pereira, M. Transcriptional profile of Paracoccidioides induced by oenothein B, a potential antifungal agent from the Brazilian Cerrado plant Eugenia uniflora. BMC Microbiol. 2013, 13, 227, doi:10.1186/1471-2180-13-227.
- Santos, G.D.; Ferri, P.H.; Santos, S.C.; Bao, S.N.; Soares, C.M.A.; Pereira, M. Oenothein B inhibits the expression of PbFKS1 transcript and induces morphological changes in Paracoccidioides brasiliensis. Med. Mycol. 2007, 45, 609–618, doi:10.1080/13693780701502108.
- do Prado, A.C.; Garces, H.G.; Bagagli, E.; Rall, V.L.M.; Furlanetto, A.; Fernandes Junior, A.; Furtado, F.B. Schinus molle essential oil as a potential source of bioactive compounds: Antifungal and antibacterial properties. J. Appl. Microbiol. 2019, 126, 516–522, doi:10.1111/jam.14157.
- Johann, S.; Cisalpino, P.S.; Watanabe, G.A.; Cota, B.B.; de Siqueira, E.P.; Pizzolatti, M.G.; Zani, C.L.; de Resende, M.A. Antifungal activity of extracts of some plants used in Brazilian traditional medicine against the pathogenic fungus Paracoccidioides brasiliensis. Pharm. Biol. 2010, 48, 388–396, doi:10.3109/13880200903150385.
- Johann, S.; Oliveira, F.B.; Siqueira, E.P.; Cisalpino, P.S.; Rosa, C.A.; Alves, T.M.A.; Zani, C.L.; Cota, B.B. Activity of compounds isolated from Baccharis dracunculifolia D.C. (Asteraceae) against Paracoccidioides brasiliensis. Med. Mycol. 2012, 50, 843–851, doi:10.3109/13693786.2012.678903.
- Neelofar, K.; Shreaz, S.; Rimple, B.; Muralidhar, S.; Nikhat, M.; Khan, L.A. Curcumin as a promising anticandidal of clinical interest. Can. J. Microbiol. 2011, 57, 204–210, doi:10.1139/W10-117.
- San-Blas, G.; Mariño, L.; San-Blas, F.; Apitz-Castro, R. Effect of ajoene on dimorphism of Paracoccidioides brasiliensis. Med. Mycol. 1993, 31, 133–141, doi:10.1080/02681219380000151.
- Mendes, G.; Baltazar, L.M.; Souza, D.G.; Sá, N.P.; Rosa, L.H.; Rosa, C.A.; Souza-Fagundes, E.M.; Ramos, J.P.; Alves-Silva, J.; Cota, B.B.; et al. Effects of cytochalasin E on Paracoccidioides brasiliensis. J. Appl. Microbiol. 2018, 125, 1296–1307, doi:10.1111/jam.14053.
- Campos, F.F.; Johann, S.; Cota, B.B.; Alves, T.M.A.; Rosa, L.H.; Caligiorne, R.B.; Cisalpino, P.S.; Rosa, C.A.; Zani, C.L. Antifungal activity of trichothecenes from Fusarium sp. against clinical isolates of Paracoccidioides brasiliensis: Antifungal activity of trichothecenes. Mycoses 2011, 54, e122–e129, doi:10.1111/j.1439-0507.2009.01854.x.
- Johann, S.; Rosa, L.H.; Rosa, C.A.; Perez, P.; Cisalpino, P.S.; Zani, C.L.; Cota, B.B. Antifungal activity of altenusin isolated from the endophytic fungus Alternaria sp. against the pathogenic fungus Paracoccidioides brasiliensis. Rev. Iberoam. Micol. 2012, 29, 205–209, doi:10.1016/j.riam.2012.02.002.
- Derengowski, L.S.; De-Souza-Silva, C.; Braz, S.V.; Mello-De-Sousa, T.M.; Báo, S.N.; Kyaw, C.M.; Silva-Pereira, I. Antimicrobial effect of farnesol, a Candida albicans quorum sensing molecule, on Paracoccidioides brasiliensis growth and morphogenesis. Ann Clin. Microbiol. Antimicrob. 2009, 8, 13, doi:10.1186/1476-0711-8-13.
- Saleem, H.; Maryam, A.; Bokhari, S.A.; Ashiq, A.; Rauf, S.A.; Khalid, R.R.; Qureshi, F.A.; Siddiqi, A.R. Design, synthesis, characterization and computational docking studies of novel sulfonamide derivatives. EXCLI J. 2018, 17, 169–180, doi:10.17179/excli2017-886.
- Chen, J.; Xie, S. Overview of sulfonamide biodegradation and the relevant pathways and microorganisms. Sci. Total Environ. 2018, 640–641, 1465–1477, doi:10.1016/j.scitotenv.2018.06.016.