People with Alzheimer’s disease (AD) have significantly higher rates of subclinical and overt epileptiform activity. In animal models, oligomeric Aβ amyloid is able to induce neuronal hyperexcitability even in the early phases of the disease. Such aberrant activity subsequently leads to downstream accumulation of toxic proteins, and ultimately to further neurodegeneration and neuronal silencing mediated by concomitant tau accumulation. Several neurotransmitters participate in the initial hyperexcitable state, with increased synaptic glutamatergic tone and decreased GABAergic inhibition. These changes appear to activate excitotoxic pathways and, ultimately, cause reduced long-term potentiation, increased long-term depression, and increased GABAergic inhibitory remodelling at the network level. Brain hyperexcitability has therefore been identified as a potential target for therapeutic interventions aimed at enhancing cognition, and, possibly, disease modification in the longer term. Clinical trials are ongoing to evaluate the potential efficacy in targeting hyperexcitability in AD, with levetiracetam showing some encouraging effects. Newer compounds and techniques, such as gene editing via viral vectors or brain stimulation, also show promise. Diagnostic challenges include identifying best biomarkers for measuring sub-clinical epileptiform discharges. Determining the timing of any intervention is critical and future trials will need to carefully stratify participants with respect to the phase of disease pathology.
- Alzheimer’s disease
- epilepsy
- hyperexcitability
- neurodegeneration
- Alzheimer’s disease,epilepsy,hyperexcitability,neurodegeneration
1. Introduction
Recent clinical and preclinical research has led to a growing realization of the strong association between brain hyperexcitability, manifest in its extreme form as epilepsy, and Alzheimer’s disease (AD)
. Epileptiform activity in AD might arise as a bystander effect, encountered as consequence of neurodegeneration as the disease progresses. On the other hand, it might be a constituent component of the AD phenotype
. It is now, for example, established that AD patients have higher rates of subclinical and overt epileptiform activity
[2]
. The prevalence of subclinical epileptiform activity is still largely unknown
[5]
, with some evidence suggesting it could be present in up to 42.4% of AD cases
[6]
. Clinically overt seizures among AD patients have been reported to be from 6 to 17 times higher compared to age-matched controls
, while the lifetime prevalence of seizures in AD populations ranges from 1.5 to 64%, partly owing to the pleomorphic clinical representations of epileptic discharges
. Most seizures are subtle and non-convulsive in AD; they could easily be missed, and confusional or amnestic episodes overlap with typical AD symptoms
.
Preclinical data in both AD and epilepsy models show that covert epileptic discharges can have an adverse impact on cognition
. Murine models of epilepsy frequently report behavioural impairment in standard tests of spatial cognition such as the Morris water maze task
, with a disruption of precise temporal organization of neuronal firing that is essential for normal cognitive processing
[18]
. Epileptiform discharges are also associated with impaired performance in cognitive tasks, usually involving memory and spatial processing in mouse models of AD
. Similarly, subclinical epileptiform activity in AD patients associates with an earlier and more rapid cognitive decline, in both memory and executive function
.
2. Who, When, and How to Treat Brain Hyperexcitability: Diagnostic and Therapeutic Challenges
Preclinical and human studies show that seizure susceptibility is higher if a genetic risk factor for early or late onset AD is present
. Young patients who carry APP, PSEN1, or PSEN2 mutations show an increased prevalence of seizures compared to sporadic AD patients
[11]
, which could be as high as 87 fold
[24]
. ApoE4+ mice show increased hyperexcitability, especially in the entorhinal cortex, even independently of Aβ and tau pathology
[25]
, implying that ApoE4 genotype might be a distinct risk factor for hyperexcitability. Young healthy humans who are ApoE4 carriers also show fMRI hyperactivity of the hippocampus
[26]
. Adeno-associated virus (AAV) vectors, and specifically the AAVrh.10-APOE2 vector, have shown promising results in mice and non-human primates in shifting the more detrimental ApoE4 genotype expression to ApoE2, with a single intracerebral injection resulting in decreased Aβ levels and amyloid plaque formation
. A pioneering phase 1 study with AAVrh.10-APOE2 vector is currently ongoing in ApoE4+ MCI and AD patients. One possible implication therefore is that ApoE4+ individuals might be an important group to target for initial attempts to reduce brain hyperexcitability, but further data in humans are needed to confirm these promising preclinical data.
2.1. Diagnostic Tools
Whereas counteracting
hyper
excitability might be the optimal strategy in early phases of AD, preventing neuronal
hypo
excitability might be crucial in later phases
[28]
. Therefore, the timing of therapeutic strategies in different stages of AD (preclinical, prodromal, moderate, severe pathology) might need to be accounted for when designing clinical trials addressing neuronal hyperexcitability.
How would it be possible to stage a patient in vivo (Figure 1)?
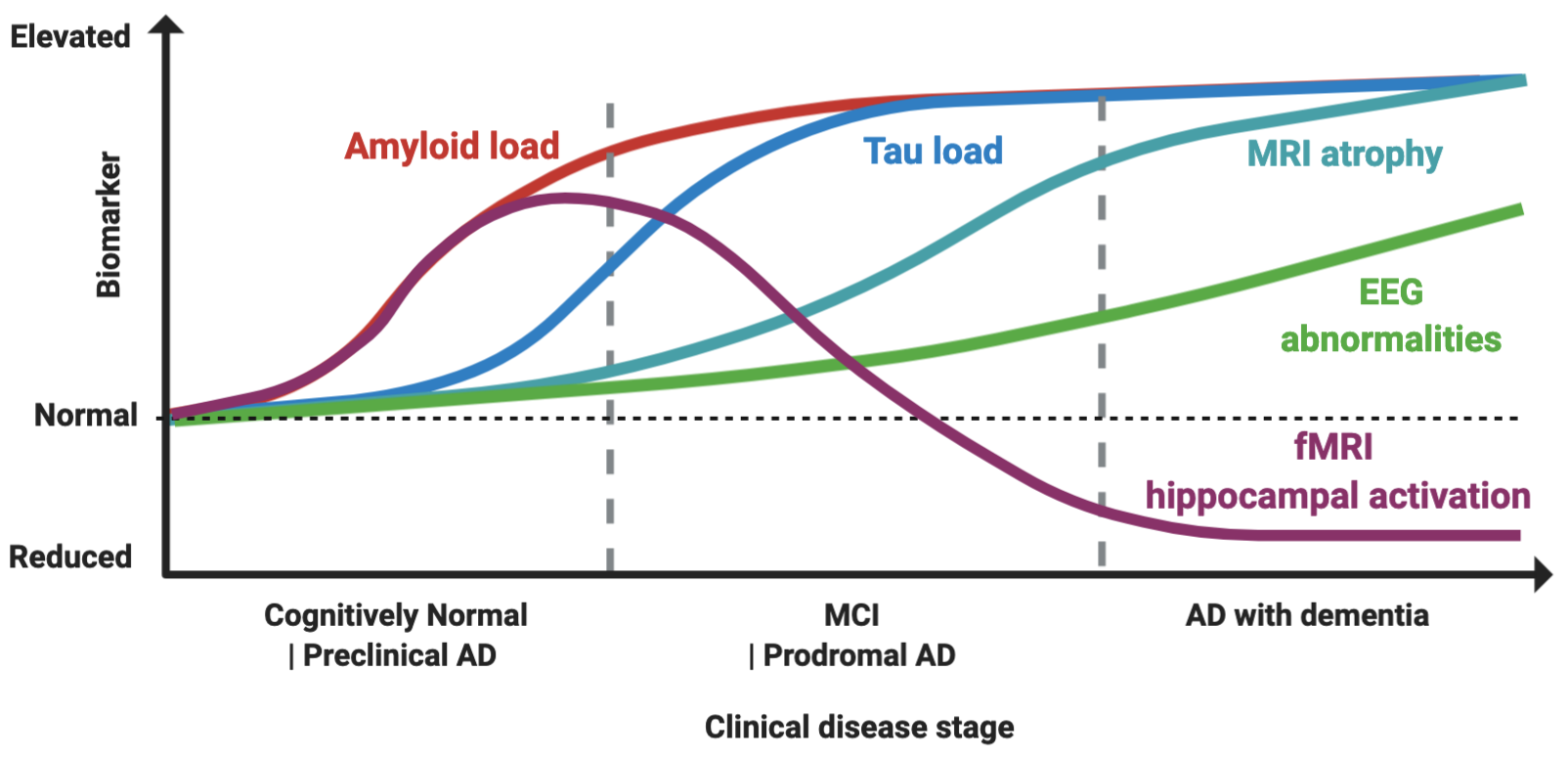
Figure 1.
Proposed model of biomarker dynamics of hyperexcitability in humans. Amyloid load, measured by either cerebrospinal fluid (CSF) or Pittsburgh B compound amyloid ligand (PiB) positron emission tomography (), is the first to increase. Functional magnetic resonance imaging (fMRI) hippocampal activation is elevated in the preclinical and early prodromal Alzheimer’s disease (AD) phases, and subsequently decreases, with final hypoactivation in AD dementia stage. Tau load elevation, at CSF analysis or tau imaging, subsequently follows. Higher rates of MRI atrophy appear after fMRI hyperactivation and tau increase. Electroencephalogram (EEG) abnormalities increase longitudinally as disease progress, with suboptimal detection rates. The combined effect of Aβ amyloid and tau induces hyperexcitability in early and hypoexcitability in late disease stages, as depicted by fMRI hippocampal activation. Made in ©BioRender - biorender.com
Hippocampal fMRI activation has gained attention as a marker of hyperexcitability, as it is increased in MCI patients compared to controls, and in early MCI compared to late MCIs, while AD patients typically show an hypoactivation pattern, thus suggesting this might reflect a temporal dynamic shift from hyper to hypoexcitability
, (
). Notably, hippocampal fMRI hyperactivation has been found also in young, cognitively-intact presymptomatic individuals with the E280A PSEN1 mutation
[33]
, in ApoE4+ individuals
[34]
, and controls with a family history of AD
[35]
, suggesting that it might be a possible signature of early preclinical neuronal dysfunction. It is also correlated with cortical thinning in brain regions typically associated with AD pathology
[36]
, to longitudinal increased amyloid accumulation measured by PiB-PET and higher rates of cognitive decline
[37]
, (
). “When” to treat seems, therefore, as soon as possible, given also that when hypoactivity is present, as shown by preclinical models, tau-related damage might already be irreversible
[38]
.
Nevertheless, task-related fMRI hyperactivity is not a direct measure of epileptiform activity, so its interpretation as marker of epileptiform activity is still speculative. One key piece of evidence strengthening this link, however, is the finding that levetiracetam is able to counteract the hippocampal hyperactivation in MCI patients
, implying that it is indeed reflecting underlying epileptiform activity.
What is the role of the most used tool to assess hyperexcitability in clinical practice, which is standard EEG? Areas of hyperexcitability might be limited to a small region such as the entorhinal cortex
, and could coexist with hypoactive circuits, even in adjacent regions
, making any changes difficult to detect by large scale surface EEG recordings
[11]
. Therefore, non-invasive scalp recording as provided by standard EEG might substantially underestimate brain hyperexcitability
[43]
. Moreover, epileptiform activity could be more prevalent during sleep
, and therefore missed in routine clinical evaluations. Even if standard EEG abnormalities, as increased theta and delta activities, have shown a potential in tracking AD progression, longitudinal EEGs as are rarely used in clinical practice for AD staging
[43]
,(
). A 24 h long-term monitoring by video-electroencephalography (LTM-EEG) telemetry has proven to increase the chances of uncovering subclinical epileptiform activity in AD patients
[5]
. Quantitative EEG (qEEG) analysis has also shown promise in detecting early neuronal dysfunction and to correlate with molecular and imaging biomarkers of the disease
[44]
. Another emerging technique to measure the disruption of neuronal fine tuning in AD is magnetoencephalography (MEG), which has several advantages over fMRI and EEG, combining high spatial and sub-millisecond temporal resolution
[45]
. MEG has been shown to outperform standard and prolonged EEG in detecting subclinical epileptiform activity in AD patients and controls
[5]
. It is able not only to detect localized patterns of reduced connectivity in AD patients
[46]
, but also to predict future conversion from MCI to AD
[47]
. MEG can detect deficits of functional connectivity even in patients with subjective cognitive impairment, possibly providing a very early maker of the disease
[48]
. Intriguingly, metrics such as Synchronization Likelihood (SL), a measure of functional connectivity, could be increased in MCI patients and reduced in AD, possibly mirroring fMRI dynamics of initial hyper and subsequent hypoactivation
[49]
.
Whether, however, these changes reflect an underlying hyperexcitable state, remains to be ascertained. Multiple MEG metrics show different trajectories alongside disease progression and MEG availability is still limited to a relatively smaller number of research centres
[50]
. One study supported the detection of Aβ-induced hyperexcitability in MCI patients, showing that Aβ-positive MCIs had increased alpha band power in medial frontal areas and increased delta band power, which correlated with disease progression within the AD continuum
[51]
. Even if some data suggest that MEG is able to record signal coming from the hippocampus, the decrease in MEG signal-to-noise ratio as a function of source depth implies that, as for surface EEG, its detection of subtle abnormalities in deep brain structures might be suboptimal
[52]
. A phase 2 clinical trial is ongoing to test the effect of levetiracetam on MEG signal changes in patients with MCI and AD. Another clinical trial (NCT04131491) is currently recruiting to quantify subclinical epileptiform discharges and hippocampal hyperactivity with MEG, prolonged EEG and their impact on CSF biomarkers of AD.
2.2. Therapeutic Tools
Different non-pharmacological brain stimulation techniques such as transcranial magnetic stimulation (TMS), transcranial direct current stimulation (tDCS), transcranial alternating current stimulation (tACS), either alone or combined with EEG have been used either to diagnose or to treat brain hyperexcitability in AD through detection and modulation of LTP and LTD changes. Given the modulatory properties of TMS, and the possibility of detecting its impact at a granular temporal scale with EEG, these techniques have also been proposed as therapeutic tools to tune the brain’s excitatory state
[53]
. TMS protocols have been extensively used in AD for diagnostic and therapeutic purposes
, particularly Theta burst Stimulation (TBS), which resembles the methods used for investigation of hippocampal plasticity
[56]
, and its metrics of LTP reduction correlate with hippocampal-type cognitive impairment in AD
[57]
. Short latency afferent inhibition (SAI), which is a measure of cholinergic pathways’ integrity, shows that AD patients have impaired LTP-like cortical plasticity, with preservation of LTD
[58]
. TMS and TMS-EEG have been able to detect hyperexcitability in early stages of AD
. TMS-EEG with stimulation of the precuneus has been reported to ameliorate memory deficits and enhance beta oscillations in prodromal AD
[61]
, and several trials in MCI or AD are currently ongoing.
Several small studies with tDCS have shown some efficacy in enhancing memory function in AD patients, even if with conflicting results
. tACS, with its ability to entrain or synchronize brain network oscillations, especially in the 40 Hz gamma frequency, is being explored as a therapeutic tool in AD disease
[66]
. GammaSense stimulation, which delivers a LED light flashing at 40 Hz and auditory stimuli, has shown promise in different mouse models, including 5XFAD, APP/PS1, and wild type mice, with reduction of Aβ and tau levels and positive effect on microglia
. Positive effects in reducing amyloid load in auditory cortex and hippocampus, as well as a more widespread reduction of Aβ load, and improved spatial and recognition memory of 5XFAD mice, have been reported
[69]
. Moreover, reduced tau phosphorylation has been found in the P301S tauopathy model after GammaSense treatment
[69]
. Human studies applying GammaSense stimulation in MCI or AD are currently ongoing, though a small pilot study in 10 patients on 40 Hz light therapy had no effects on Aβ load
[70]
. Some groups have also coupled TMS or tDCS with cognitive stimulation
[71]
,
[72]
. Other devices, such as NeuroEM, based on Transcranial Electromagnetic Treatment (TEMT), seem to show promising results
[73]
, and clinical trials to assess its efficacy are currently ongoing. Alternative approaches are also being studied, such as temporal interference stimulation (TI), which can selectively modulate neurons in the deep brain structures in animal models and human prototypes
. Intranasal delivery of near infrared (NIR) light via light emitting diodes, or photobiomodulation is also being tested in AD for its possible beneficial impact of mitochondrial function, and improvements in cognition, increased cerebral perfusion, and enhanced connectivity between the posterior cingulate cortex and lateral parietal nodes of the default-mode network after 12 weeks of treatment have been reported in a small pilot study
[76]
.
All of these non-pharmacological brain stimulation techniques have their limitations. Some of these stimulation protocols have “history of seizure” as exclusion criterion, as they can lower seizure threshold
[76]
, which might be extremely important in the context of increased hyperexcitability in AD patients. Moreover, the reported positive effects on cognition usually last only for few weeks after stimulation, and there is still little evidence for long-term cognitive benefit
[77]
. Besides these new approaches, which are available in the context of research, different pharmacological compounds such as anti-seizure medications (ASMs) have been used to address the question of “How” to treat brain hyperexcitability, targeting different steps of the excitotoxic cascade (
), and they remain at the moment the most reliable option.
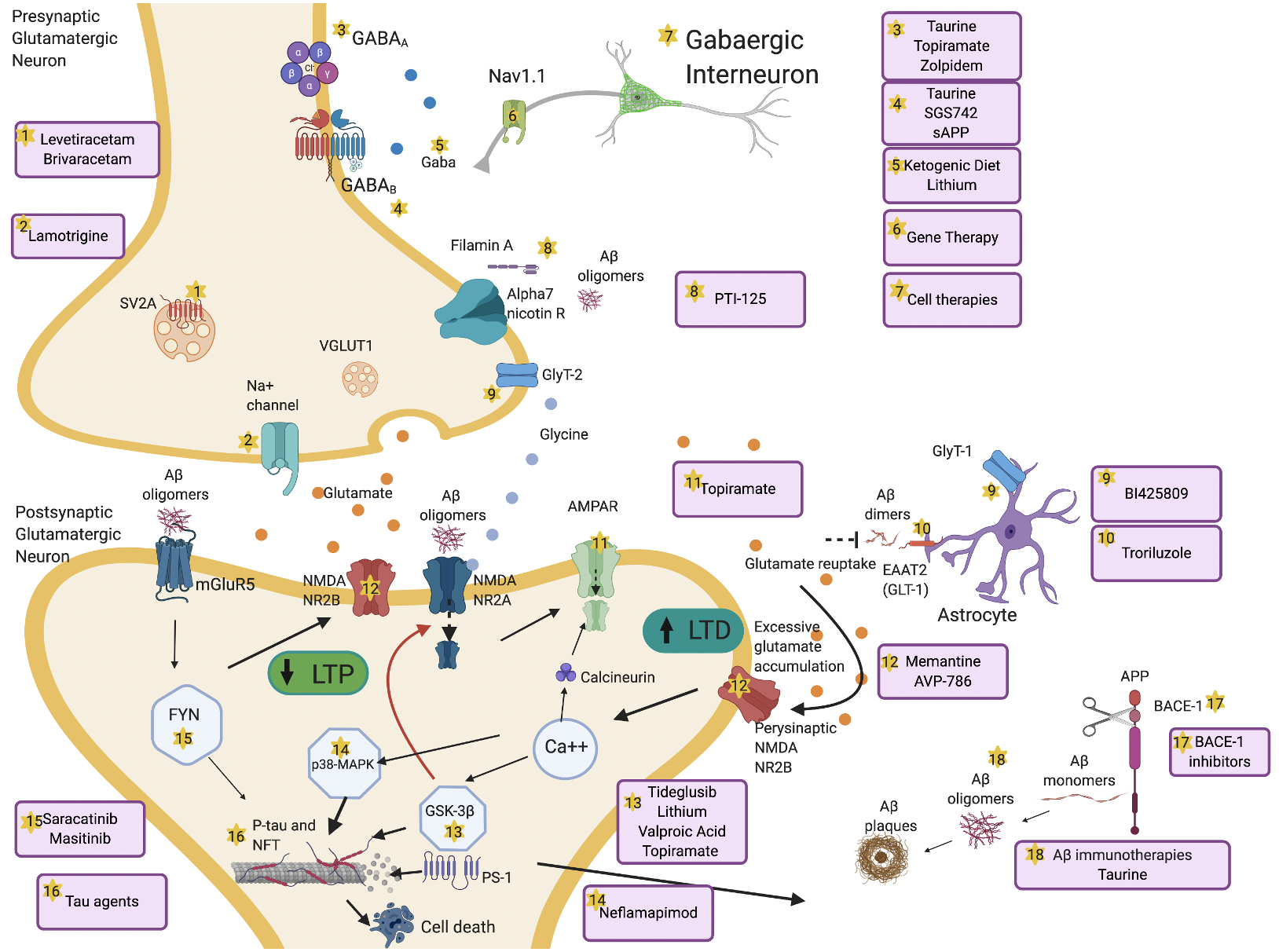
Figure 2. Overview of mechanisms and therapeutic targets of hyperexcitability in AD. Aβ dimers block glutamate reuptake by astrocytes through glutamate transporter-1 (GLT-1) receptors. This causes increased glutamate levels in the synaptic cleft, activation of perisynaptic N-methyl-D-aspartate (NMDA) 2B receptors, increased Ca++ influx, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors internalization and activation of glycogen synthase kinase 3 beta (GSK-3β) and p38 mitogen-activated protein kinase (p38-MAPK) pathways. These pathologic cascades lead to abnormal tau phosphorylation and neurodegeneration. Long-term potentiation (LTP) is reduced and long-term depression (LTD) increases. Aβ oligomers interact pre- and postsynaptically with alpha-7 nicotinic receptors (alpha7-nAChRs), metabotropic glutamate receptors 5 (mGluR5s), and NMDA receptors. mGluR5 activates Fyn-mediated neurodegenerative changes. Increased excitation can also be driven by presynaptic changes in synaptic vesicle glycoprotein (SV2A) and Na+ channels. Decrease of GABAergic transmission or impaired glycine levels are also implicated in increasing hyperexcitability. Several therapeutic compounds are able to counteract specific molecular targets implicated in hyperexcitability. Made in ©BioRender - biorender.com.
3. Conclusions
Hyperexcitability, especially localized to the hippocampus, seems to be an early signature of neuronal and cognitive dysfunction in patients who are at risk of developing AD
. Preclinical models and human studies suggest that these changes reflect an early aberrant E > I (excitatory > inhibitory) imbalance, which is associated with Aβ synaptopathy, and fosters further reactive release of toxic compounds such as Aβ amyloid and tau
. These alterations might decrease during disease progression, as shown by the progressive tau induced neuronal silencing, i.e., E < I, and subsequent neurodegeneration in the later phases of the disease
. Therefore, there might be a very narrow window of opportunity to target brain hyperexcitability, which might need to be taken into account when designing clinical trials tackling hyperexcitability in AD.
Several ASMs have been proposed as a means of counteracting brain hyperexcitability in preclinical models of AD, as well as in patients
[81]
, with levetiracetam showing promising results
[82]
. GABAergic modulation is also being explored, through repurposing of licensed medications; new GABA
A
agonists and GABA
B
antagonists; and innovative techniques such as gene and stem cell therapies
[83]
.
Targeting cardiovascular risk factors, such as hypertension and diabetes, has been proposed to counteract the development of additional vascular lesions in AD patients, but also to help reduce brain hyperexcitability
[84]
. Clinical trials to tackle neuroinflammation, rather than systemic inflammation, through more tailored approaches are ongoing, as is work on gene editing via viral vectors to reduce the detrimental and pro-excitatory effects of ApoE4 genotype
[26]
.
Non-pharmacological stimulation techniques have also been shown to enhance cognition in AD patients, at least in the short-term, by modulating brain hyperexcitability, and are being trialed for their possible long-term effects on AD pathological cascades.
One of the critical questions is what defines the best in vivo marker for hyperexcitability, as this would help stratify people with AD for clinical trials. In humans, fMRI has shown promising results in detecting early hippocampal alterations
, but other approaches such as MEG or TMS-EEG might also be considered to measure brain hyperexcitability owing to their good temporal resolution and modulation potential
.
Clinical trials targeting different molecular pathways that contribute to the genesis of such aberrant cortical function, as well as being of therapeutic relevance, offer insights on AD progression and how to potentially prevent the development of dementia in susceptible populations. Nevertheless, several clinical trials have failed so far in halting AD progression through modulation of possible targets of brain hyperexcitability, and multiple diagnostic and therapeutic challenges have yet to be overcome. Licensed drugs, as well as new strategies are being tested in cognitively healthy people at risk of developing AD, as well as in MCI and AD patients, mostly in early-prodromal phases. These upcoming trials could cast a light on the potential of brain fine-tuning, and possible disease modifying effects in AD.
References
- Samuel S. Harris; Fred Wolf; Bart De Strooper; Marc A. Busche; Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer’s Disease. Neuron 2020, 107, 417-435, 10.1016/j.neuron.2020.06.005.
- Jorge J. Palop; Epilepsy and Cognitive Impairments in Alzheimer Disease. Archives of Neurology 2009, 66, 435-440, 10.1001/archneurol.2009.15.
- Keith A Vossel; Maria C Tartaglia; Haakon B Nygaard; Adam Z Zeman; Bruce L Miller; Epileptic activity in Alzheimer's disease: causes and clinical relevance. The Lancet Neurology 2017, 16, 311-322, 10.1016/s1474-4422(17)30044-3.
- Marc Edwards; N P Robertson; Seizures in Alzheimer’s disease: is there more beneath the surface?. Journal of Neurology 2017, 265, 226-228, 10.1007/s00415-017-8694-6.
- Keith A. Vossel; Kamalini G. Ranasinghe; Alexander J. Beagle Ba; Danielle Mizuiri Bs; Susanne M. Honma Bs; Anne F. Dowling Ms; Sonja M. Darwish Ms; Victoria Van Berlo Bs; Deborah E. Barnes; Mary Mantle; et al.Anna M. Karydas BaGiovanni CoppolaErik D. Roberson MdBruce L. MillerPaul A. GarciaHeidi E. KirschLennart MuckeSrikantan S. Nagarajan Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Annals of Neurology 2016, 80, 858-870, 10.1002/ana.24794.
- Dale C. Hesdorffer; W. A. Hauser; J. F. Annegers; E. Kokmen; W. A. Rocca; Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727-730, 10.1212/wnl.46.3.727.
- W. A. Hauser; M. L. Morris; L. L. Heston; V. E. Anderson; Seizures and myoclonus in patients with Alzheimer's disease. Neurology 1986, 36, 1226-1226, 10.1212/wnl.36.9.1226.
- Jacopo C. DiFrancesco; Lucio Tremolizzo; Valeria Polonia; Giorgia Giussani; Elisa Bianchi; Carlotta Franchi; Alessandro Nobili; Ildebrando Appollonio; Ettore Beghi; Carlo Ferrarese; et al. Adult-Onset Epilepsy in Presymptomatic Alzheimer’s Disease: A Retrospective Study. Journal of Alzheimer's Disease 2017, 60, 1267-1274, 10.3233/jad-170392.
- Dionysios Pandis; Nikolaos Scarmeas; Seizures in Alzheimer Disease: Clinical and Epidemiological Data. Epilepsy Currents 2012, 12, 184-187, 10.5698/1535-7511-12.5.184.
- Nikolaos Scarmeas; Lawrence S. Honig; Hyunmi Choi; Julio Cantero; Jason Brandt; Deborah Blacker; Marilyn Albert; Joan C. Amatniek; Karen Marder; Karen Bell; et al.W. Allen HauserYaakov Stern Seizures in Alzheimer Disease. Archives of Neurology 2009, 66, 992-997, 10.1001/archneurol.2009.130.
- Alice D. Lam; Gina Deck; Alica Goldman; Emad N. Eskandar; Jeffrey Noebels; Andrew J. Cole; Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nature Medicine 2017, 23, 678-680, 10.1038/nm.4330.
- John Baker; Tina Libretto; William Henley; Adam Zeman; The prevalence and clinical features of epileptic seizures in a memory clinic population. Seizure 2019, 71, 83-92, 10.1016/j.seizure.2019.06.016.
- Jorge J. Palop; Jeannie Chin; Erik D. Roberson; Jun Wang; Myo T. Thwin; Nga Bien-Ly; Jong Yoo; Kaitlyn O. Ho; Gui-Qiu Yu; Anatol Kreitzer; et al.Steven FinkbeinerJeffrey L. NoebelsLennart Mucke Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer's Disease. Neuron 2007, 55, 697-711, 10.1016/j.neuron.2007.07.025.
- Ludmyla Kandratavicius; Priscila Alves Balista; Cleiton Lopes-Aguiar; Rafael Naime Ruggiero; Eduardo Henrique Umeoka; Norberto Garcia-Cairasco; Lezio Soares Bueno-Junior; Joao Pereira Leite; Animal models of epilepsy: use and limitations. Neuropsychiatric Disease and Treatment 2014, 10, 1693-705, 10.2147/ndt.s50371.
- Xianzeng Liu; Robert U. Muller; Li-Tung Huang; John L. Kubie; Alexander Rotenberg; Bruno Rivard; Maria Roberta Cilio; Gregory L. Holmes; Seizure-Induced Changes in Place Cell Physiology: Relationship to Spatial Memory. Journal of Neuroscience 2003, 23, 11505-11515, 10.1523/jneurosci.23-37-11505.2003.
- Tristan Shuman; D. Aharoni; Denise J. Cai; Christopher R. Lee; Spyridon Chavlis; Lucia Page-Harley; Lauren M. Vetere; Yu Feng; Chen Yi Yang; Irene Mollinedo-Gajate; et al.Lingxuan ChenZachary T. PenningtonJiannis TaxidisSergio E. FloresKevin ChengMilad JavaherianChristina C. KabaNaina RaoMimi La-VuIoanna PandiMatthew ShtrahmanKonstantin I. BakhurinSotiris C. MasmanidisBaljit S. KhakhPanayiota PoiraziAlcino J. SilvaPeyman Golshani Breakdown of spatial coding and interneuron synchronization in epileptic mice.. Nature Neuroscience 2020, 23, 229-238, 10.1038/s41593-019-0559-0.
- Gregory L. Holmes; Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disorders 2015, 17, 101-116, 10.1684/epd.2015.0739.
- Erik D. Roberson; Kimberly Scearce-Levie; Jorge J. Palop; Fengrong Yan; Irene H. Cheng; Tiffany Wu; Hilary Gerstein; Gui-Qiu Yu; Lennart Mucke; Reducing Endogenous Tau Ameliorates Amyloid -Induced Deficits in an Alzheimer's Disease Mouse Model. Science 2007, 316, 750-754, 10.1126/science.1141736.
- Rimante Minkeviciene; Sylvain Rheims; Marton B. Dobszay; Misha Zilberter; Jarmo Hartikainen; Lívia Fülöp; Botond Penke; Yuri Zilberter; Tibor Harkany; Asla Pitkänen; et al.Heikki Tanila Amyloid -Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. Journal of Neuroscience 2009, 29, 3453-3462, 10.1523/jneurosci.5215-08.2009.
- Jorge J. Palop; Lennart Mucke; Amyloid-β–induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nature Neuroscience 2010, 13, 812-818, 10.1038/nn.2583.
- Yaisa Andrews-Zwilling; Nga Bien-Ly; Qin Xu; Gang Li; Aubrey Bernardo; Seo Yeon Yoon; Daniel Zwilling; Tonya Xue Yan; Ligong Chen; Yadong Huang; et al. Apolipoprotein E4 Causes Age- and Tau-Dependent Impairment of GABAergic Interneurons, Leading to Learning and Memory Deficits in Mice. Journal of Neuroscience 2010, 30, 13707-13717, 10.1523/jneurosci.4040-10.2010.
- Orwa Aboud Sue T Griffin W; Robert E. Mrak; Frederick Boop; Sue T Griffin; Epilepsy: neuroinflammation, neurodegeneration, and APOE genotype. Acta Neuropathologica Communications 2013, 1, 41-41, 10.1186/2051-5960-1-41.
- Joan C. Amatniek; W. Allen Hauser; Carrie DelCastillo-Castaneda; Diane M. Jacobs; Karen Marder; Karen Bell; Marilyn Albert; Joseph Brandt; Yaakov Stern; Incidence and Predictors of Seizures in Patients with Alzheimer's Disease. Epilepsia 2006, 47, 867-872, 10.1111/j.1528-1167.2006.00554.x.
- Tal Nuriel; Sergio L. Angulo; Usman Khan; Archana Ashok; Qiuying Chen; Helen Y. Figueroa; Sheina Emrani; Li Liu; Mathieu Herman; Geoffrey M. Barrett; et al.Valerie SavageLuna BuitragoEfrain Cepeda-PradoChristine FungEliana GoldbergSteven S. GrossS. Abid HussainiHerman MorenoScott A SmallKaren E. Duff Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nature Communications 2017, 8, 1464, 10.1038/s41467-017-01444-0.
- Lukas Kunz; Tobias Navarro Schröder; HweeLing Lee; Christian Montag; Bernd Lachmann; Rayna Sariyska; Martin Reuter; Rüdiger Stirnberg; Tony Stöcker; Paul Christian Messing-Floeter; et al.Juergen FellChristian F. DoellerNikolai Axmacher Reduced grid-cell-like representations in adults at genetic risk for Alzheimer's disease. Science 2015, 350, 430-433, 10.1126/science.aac8128.
- Lingzhi Zhao; Andrew J. Gottesdiener; Mayur Parmar; Mingjie Li; Stephen M. Kaminsky; Maria J. Chiuchiolo; Dolan Sondhi; Patrick M. Sullivan; David M. Holtzman; Ronald G. Crystal; et al.Steven M. Paul Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer's disease mouse models. Neurobiology of Aging 2016, 44, 159-172, 10.1016/j.neurobiolaging.2016.04.020.
- Jonathan B. Rosenberg; Michael G. Kaplitt; Bishnu P. De; Alvin Chen; Thomas Flagiello; Christiana O Salami; Eduard Pey; Lingzhi Zhao; Rodolfo J. Ricart Arbona; Sebastien Monette; et al.Jonathan P. DykeDouglas J. BallonStephen M. KaminskyDolan SondhiGregory A. PetskoSteven M. PaulRonald G. Crystal AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer's Disease. Human Gene Therapy Clinical Development 2018, 29, 24-47, 10.1089/humc.2017.231.
- Arjune Sen; Valentina Capelli; Masud Husain; Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592-1608, 10.1093/brain/awy022.
- B. C. Dickerson; D. H. Salat; D. N. Greve; E. F. Chua; E. Rand-Giovannetti; D. M. Rentz; L. Bertram; K. Mullin; R. E. Tanzi; D. Blacker; et al.M. S. AlbertR. A. Sperling Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005, 65, 404-411, 10.1212/01.wnl.0000171450.97464.49.
- Anne Hämäläinen; Maija Pihlajamäki; Heikki Tanila; Tuomo Hänninen; Eini Niskanen; Susanna Tervo; Pasi A. Karjalainen; Ritva L. Vanninen; Hilkka Soininen; Increased fMRI responses during encoding in mild cognitive impairment. Neurobiology of Aging 2007, 28, 1889-1903, 10.1016/j.neurobiolaging.2006.08.008.
- Michael A. Yassa; Shauna M. Stark; Arnold Bakker; Marilyn S. Albert; Michela Gallagher; Craig E. L. Stark; High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage 2010, 51, 1242-1252, 10.1016/j.neuroimage.2010.03.040.
- Kim A. Celone; Vince D. Calhoun; Bradford C. Dickerson; Alireza Atri; Elizabeth F. Chua; Saul L. Miller; Kristina DePeau; Doreen M. Rentz; Dennis J. Selkoe; Deborah Blacker; et al.Marilyn S. AlbertReisa A Sperling Alterations in Memory Networks in Mild Cognitive Impairment and Alzheimer's Disease: An Independent Component Analysis. Journal of Neuroscience 2006, 26, 10222-10231, 10.1523/jneurosci.2250-06.2006.
- Yakeel T. Quiroz Ma; Andrew E. Budson; Kim Celone; Adriana Ruiz; Randall Newmark Ba; Gabriel Castrillón Bs; Francisco Lopera; Chantal E. Stern; Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Annals of Neurology 2010, 68, 865-875, 10.1002/ana.22105.
- Mark W. Bondi; Wes S. Houston; Lisa T. Eyler; Gregory G. Brown; fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 2005, 64, 501-508, 10.1212/01.wnl.0000150885.00929.7e.
- Susan Spear Bassett; David M. Yousem; Catherine Cristinzio; Ivana Kusevic; Michael A. Yassa; Brian S. Caffo; Scott L. Zeger; Familial risk for Alzheimer's disease alters fMRI activation patterns. Brain 2006, 129, 1229-1239, 10.1093/brain/awl089.
- Deepti Putcha; Michael Brickhouse; Kelly O'keefe; Caroline Sullivan; Dorene Rentz; Gad Marshall; Brad Dickerson; Reisa A Sperling; Hippocampal Hyperactivation Associated with Cortical Thinning in Alzheimer's Disease Signature Regions in Non-Demented Elderly Adults. Journal of Neuroscience 2011, 31, 17680-17688, 10.1523/jneurosci.4740-11.2011.
- Willem Huijbers; Elizabeth C. Mormino; Aaron P. Schultz; Sarah Wigman; Andrew M. Ward; Mykol Larvie; Rebecca E. Amariglio; Gad A. Marshall; Dorene M. Rentz; Keith A. Johnson; et al.Reisa A. Sperling Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 2015, 138, 1023-1035, 10.1093/brain/awv007.
- Arnold Bakker; Gregory L. Krauss; Marilyn S. Albert; Caroline L. Speck; Lauren R. Jones; Craig E. Stark; Michael A. Yassa; Susan S. Bassett; Amy L. Shelton; Michela Gallagher; et al. Reduction of Hippocampal Hyperactivity Improves Cognition in Amnestic Mild Cognitive Impairment. Neuron 2012, 74, 467-474, 10.1016/j.neuron.2012.03.023.
- Arnold Bakker; Marilyn S. Albert; Gregory Krauss; Caroline L. Speck; Michela Gallagher; Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage: Clinical 2015, 7, 688-698, 10.1016/j.nicl.2015.02.009.
- Sofya Ziyatdinova; Kestutis Gurevicius; Nino Kutchiashvili; Tamuna Bolkvadze; Jari Nissinen; Heikki Tanila; Asla Pitkänen; Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Research 2011, 94, 75-85, 10.1016/j.eplepsyres.2011.01.003.
- Keith A. Vossel; Alexander J. Beagle; Gil D. Rabinovici; Huidy Shu; Suzee E. Lee; Georges Naasan; Manu Hegde; Susannah B. Cornes; Maya L. Henry; Alexandra B. Nelson; et al.William W. SeeleyMichael D. GeschwindMaria L. Gorno-TempiniTina ShihHeidi E. KirschPaul A. GarciaBruce L. MillerLennart Mucke Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurology 2013, 70, 1158-1166, 10.1001/jamaneurol.2013.136.
- Marc A. Busche; Susanne Wegmann; Simon Dujardin; Caitlin Commins; Julia Schiantarelli; Naomi Klickstein; Tarun V. Kamath; George A. Carlson; Israel Nelken; Bradley T. Hyman; et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nature Neuroscience 2018, 22, 57-64, 10.1038/s41593-018-0289-8.
- Jaeseung Jeong; EEG dynamics in patients with Alzheimer's disease. Clinical Neurophysiology 2004, 115, 1490-1505, 10.1016/j.clinph.2004.01.001.
- Una Smailovic; Vesna Jelic; Neurophysiological Markers of Alzheimer’s Disease: Quantitative EEG Approach. Neurology and Therapy 2019, 8, 37-55, 10.1007/s40120-019-00169-0.
- Edward Zamrini; Fernando Maestu; Eero Pekkonen; Michael Funke; Jyrki Makela; Myles Riley; Ricardo Bajo; Gustavo Sudre; Alberto Fernández; Nazareth Castellanos; et al.Francisco Del PozoC. J. StamBob W. Van DijkAnto BagicJames T. Becker Magnetoencephalography as a Putative Biomarker for Alzheimer's Disease. International Journal of Alzheimer's Disease 2011, 2011, 1-10, 10.4061/2011/280289.
- Cornelis J Stam; B.F. Jones; I. Manshanden; Anne-Marie Van Cappellen Van Walsum; T. Montez; J.P.A. Verbunt; J.C. De Munck; B.W. Van Dijk; H.W. Berendse; P. Scheltens; et al. Magnetoencephalographic evaluation of resting-state functional connectivity in Alzheimer's disease. NeuroImage 2006, 32, 1335-1344, 10.1016/j.neuroimage.2006.05.033.
- Fernando Maestú; Pablo Campo; P. Gil-Gregorio; S. Fernandez; T. Ortíz; Alberto Fernández; Medial temporal lobe neuromagnetic hypoactivation and risk for developing cognitive decline in elderly population: A 2-year follow-up study. Neurobiology of Aging 2006, 27, 32-37, 10.1016/j.neurobiolaging.2005.01.005.
- D. López-Sanz; R. Bruña; P. Garcés; C. Camara; N. Serrano; I. C. Rodríguez-Rojo; M. L. Delgado; M. Montenegro; R. López-Higes; M. Yus; et al.F. Maestú Alpha band disruption in the AD-continuum starts in the Subjective Cognitive Decline stage: a MEG study. Scientific Reports 2016, 6, 37685, 10.1038/srep37685.
- Ricardo Bajo; Fernando Maestú; Angel Nevado; Miguel Sancho; Ricardo Gutiérrez; Pablo Campo; Nazareth P. Castellanos; Pedro Gil; Stephan Moratti; Ernesto Pereda; et al.Francisco Del-Pozo Functional Connectivity in Mild Cognitive Impairment During a Memory Task: Implications for the Disconnection Hypothesis. Journal of Alzheimer's Disease 2010, 22, 183-193, 10.3233/jad-2010-100177.
- Pravat K. Mandal; Anwesha Banerjee; Manjari Tripathi; Ankita Sharma; A Comprehensive Review of Magnetoencephalography (MEG) Studies for Brain Functionality in Healthy Aging and Alzheimer's Disease (AD). Frontiers in Computational Neuroscience 2018, 12, 60, 10.3389/fncom.2018.00060.
- Akinori Nakamura; Pablo Cuesta; Alberto Fernández; Yutaka Arahata; Kaori Iwata; Izumi Kuratsubo; Masahiko Bundo; Hideyuki Hattori; Takashi Sakurai; Koji Fukuda; et al.Yukihiko WashimiHidetoshi EndoAkinori TakedaKersten DiersRicardo BajoFernando MaestúKengo ItoTakashi Kato Electromagnetic signatures of the preclinical and prodromal stages of Alzheimer’s disease. Brain 2018, 141, 1470-1485, 10.1093/brain/awy044.
- Emily Ruzich; Maité Crespo‐García; Sarang S. Dalal; Justin Schneiderman; Characterizing hippocampal dynamics with MEG: A systematic review and evidence‐based guidelines. Human Brain Mapping 2018, 40, 1353-1375, 10.1002/hbm.24445.
- Chun-Hung Chang; Hsien-Yuan Lane; Chieh-Hsin Lin; Brain Stimulation in Alzheimer's Disease. Frontiers in Psychiatry 2018, 9, 201, 10.3389/fpsyt.2018.00201.
- Francesco Di Lorenzo; Caterina Motta; Elias Paolo Casula; Sonia Bonnì; Martina Assogna; Carlo Caltagirone; Alessandro Martorana; Giacomo Koch; LTP-like cortical plasticity predicts conversion to dementia in patients with memory impairment. Brain Stimulation 2020, 13, 1175-1182, 10.1016/j.brs.2020.05.013.
- Ying-Hui Chou; Viet Ton That; Mark Sundman; A systematic review and meta-analysis of rTMS effects on cognitive enhancement in mild cognitive impairment and Alzheimer's disease. Neurobiology of Aging 2020, 86, 1-10, 10.1016/j.neurobiolaging.2019.08.020.
- Ying-Zu Huang; Mark J. Edwards; Elisabeth Rounis; Kailash P. Bhatia; John C. Rothwell Md; Theta Burst Stimulation of the Human Motor Cortex. Neuron 2005, 45, 201-206, 10.1016/j.neuron.2004.12.033.
- Francesco Di Lorenzo; Caterina Motta; Sonia Bonnì; Nicola Biagio Mercuri; Carlo Caltagirone; Alessandro Martorana; Giacomo Koch; LTP-like cortical plasticity is associated with verbal memory impairment in Alzheimer's disease patients. Brain Stimulation 2019, 12, 148-151, 10.1016/j.brs.2018.10.009.
- Francesco Di Lorenzo; Viviana Ponzo; Sonia Bonnì; Caterina Motta; Priscilla C. Negrão Serra; Marco Bozzali; Carlo Caltagirone; Alessandro Martorana; Giacomo Koch; Long-term potentiation-like cortical plasticity is disrupted in Alzheimer's disease patients independently from age of onset. Annals of Neurology 2016, 80, 202-210, 10.1002/ana.24695.
- Florinda Ferreri; Fabrizio Vecchio; Luca Vollero; Andrea Guerra; Sara Petrichella; David Ponzo; Sara Määttä; Esa Mervaala; Mervi Könönen; Francesca Ursini; et al.Patrizio PasqualettiGiulio IannelloPaolo Maria RossiniVincenzo Di Lazzaro Sensorimotor cortex excitability and connectivity in Alzheimer's disease: A TMS-EEG Co-registration study. Human Brain Mapping 2016, 37, 2083-2096, 10.1002/hbm.23158.
- Giovanni Pennisi; Raffaele Ferri; Giuseppe Lanza; Mariagiovanna Cantone; Manuela Pennisi; Valentina Puglisi; Giulia Malaguarnera; Rita Bella; Transcranial magnetic stimulation in Alzheimer’s disease: a neurophysiological marker of cortical hyperexcitability. Journal of Neural Transmission 2011, 118, 587-598, 10.1007/s00702-010-0554-9.
- Giacomo Koch; Sonia Bonnì; Maria Concetta Pellicciari; Elias P. Casula; Matteo Mancini; Romina Esposito; Viviana Ponzo; Silvia Picazio; Francesco Di Lorenzo; Laura Serra; et al.Caterina MottaMichele MaiellaCamillo MarraMara CercignaniAlessandro MartoranaCarlo CaltagironeMarco Bozzali Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer's disease. NeuroImage 2018, 169, 302-311, 10.1016/j.neuroimage.2017.12.048.
- R. Ferrucci; F. Mameli; I. Guidi; S. Mrakic-Sposta; M. Vergari; S. Marceglia; F. Cogiamanian; S. Barbieri; E. Scarpini; A. Priori; et al. Transcranial direct current stimulation improves recognition memory in Alzheimer disease. Neurology 2008, 71, 493-498, 10.1212/01.wnl.0000317060.43722.a3.
- Paulo Sérgio Boggio; L P Khoury; D C S Martins; O E M S Martins; E C De Macedo; F Fregni; Temporal cortex direct current stimulation enhances performance on a visual recognition memory task in Alzheimer disease. Journal of Neurology, Neurosurgery & Psychiatry 2008, 80, 444-447, 10.1136/jnnp.2007.141853.
- E. M. Khedr; Nageh F. El Gamal; Noha Abo El-Fetoh; Hosam Khalifa; Elham M. Ahmed; Anwer M. Ali; Mostafa Noaman; Ahmed Abd El-Baki; Ahmed A. Karim; A Double-Blind Randomized Clinical Trial on the Efficacy of Cortical Direct Current Stimulation for the Treatment of Alzheimer’s Disease. Frontiers in Aging Neuroscience 2014, 6, 275, 10.3389/fnagi.2014.00275.
- Martin Bystad; Ole Grønli; Ingrid Daae Rasmussen; Nina Gundersen; Lene Nordvang; Henrik Wang-Iversen; P. M. Aslaksen; Transcranial direct current stimulation as a memory enhancer in patients with Alzheimer’s disease: a randomized, placebo-controlled trial. Alzheimer's Research & Therapy 2016, 8, 1-7, 10.1186/s13195-016-0180-3.
- Yi Xing; Penghu Wei; Changming Wang; Yi Shan; Yueying Yu; Yuchen Qiao; Beijia Xie; Xinrui Shi; Zhongfang Zhu; Jie Lu; et al.Guoguang ZhaoJianping JiaY. Tang TRanscranial AlterNating current Stimulation FOR patients with Mild Alzheimer's Disease (TRANSFORM‐AD study): Protocol for a randomized controlled clinical trial. Alzheimer's & Dementia: Translational Research & Clinical Interventions 2020, 6, 1-8, 10.1002/trc2.12005.
- Hannah Frances Iaccarino; Annabelle C. Singer; Anthony J. Martorell; Andrii Rudenko; Fan Gao; Tyler Z. Gillingham; Hansruedi Mathys; Jinsoo Seo; Oleg Kritskiy; Fatema Abdurrob; et al.Chinnakkaruppan AdaikkanRebecca Gail CanterRichard RuedaEmery Neal BrownAnnabelle C. Singer Edward S. BoydenHannah F. Iaccarino Annabelle C. Singer Anthony J. Martorell Andrii Rudenko Fan Gao Tyler Z. Gillingham Hansruedi Mathys Jinsoo Seo Oleg Kritskiy Fatema Abdurrob Chinnakkaruppan Adaikkan Rebecca G. Canter Richard Rueda Emery N. Brown Edward S. Boyden Li-H Tsai Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature Cell Biology 2016, 540, 230-235, 10.1038/nature20587.
- Chinnakkaruppan Adaikkan; Steven J. Middleton; Asaf Marco; Ping-Chieh Pao; Hansruedi Mathys; David Nam-Woo Kim; Fan Gao; Jennie Z. Young; Ho-Jun Suk; Edward S. Boyden; et al.Thomas J. McHughLi‐Huei Tsai Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929-943.e8, 10.1016/j.neuron.2019.04.011.
- Anthony J. Martorell; Abigail L. Paulson; Ho-Jun Suk; Fatema Abdurrob; Gabrielle T. Drummond; Webster Guan; Jennie Z. Young; David Nam-Woo Kim; Oleg Kritskiy; Scarlett J. Barker; et al.Vamsi MangenaStephanie M. PrinceEmery N. BrownKwanghun ChungEdward S. BoydenAnnabelle C. SingerLi‐Huei Tsai Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256-271.e22, 10.1016/j.cell.2019.02.014.
- Rola Ismail; Allan K. Hansen; Peter Parbo; Hans Brændgaard; Hanne Gottrup; David J. Brooks; Per Borghammer; The Effect of 40-Hz Light Therapy on Amyloid Load in Patients with Prodromal and Clinical Alzheimer’s Disease. International Journal of Alzheimer's Disease 2018, 2018, 1-5, 10.1155/2018/6852303.
- Marwan Sabbagh; Carl Sadowsky; Babak Tousi; Marc E. Agronin; Gustavo Alva; Carmel Armon; Charles Bernick; Andrew P. Keegan; Stella Karantzoulis; Eyal Baror; et al.Moran PloznikAlvaro Pascual-Leone Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training intervention in patients with Alzheimer's disease. Alzheimer's & Dementia 2020, 16, 641-650, 10.1016/j.jalz.2019.08.197.
- Suellen Marinho Andrade; Eliane Araújo De Oliveira; Nelson Torro Alves; Ana Cristina Gomes Dos Santos; Camila Teresa Ponce Leon De Mendonça; Danielle Dorand Amorim Sampaio; Edyllaine Elidy Querino Cavalcante Da Silva; Égina Karoline Gonçalves Da Fonsêca; Evelyn Thais De Almeida Rodrigues; Gabriela Nayara Siqueira De Lima; et al.Jamerson CarvalhoJessyca Alves Silvestre Da SilvaManuella ToledoMarine Raquel Diniz Da RosaMarcia Queiroz De Carvalho GomesMelquisedek Monteiro De OliveiraMoema Teixeira Maia LemosNágylla Gomes LimaPenha InácioPetra Maria Da Cruz Ribeiro E RodriguesRayssa Gabriela Dantas FerreiraRenata CavalcanteRenata Emanuela Lyra De Brito AranhaRegina NevesRodrigo Marmo Da Costa E SouzaThainá Magalhães PortugalWanessa Kallyne Nascimento MartinsVivian PontesThiago Monteiro De Paiva FernandesIsrael ContadorBernardino Fernández-Calvo Neurostimulation Combined With Cognitive Intervention in Alzheimer’s Disease (NeuroAD): Study Protocol of Double-Blind, Randomized, Factorial Clinical Trial. Frontiers in Aging Neuroscience 2018, 10, 334, 10.3389/fnagi.2018.00334.
- Gary Arendash; Chuanhai Cao; Haitham Abulaban; Rob Baranowski; Gary Wisniewski; Lino Becerra; Ross Andel; Xiaoyang Lin; Xiaolin Zhang; David Wittwer; et al.Jay MoultonJohn ArringtonAmanda Smith A Clinical Trial of Transcranial Electromagnetic Treatment in Alzheimer’s Disease: Cognitive Enhancement and Associated Changes in Cerebrospinal Fluid, Blood, and Brain Imaging. Journal of Alzheimer's Disease 2019, 71, 57-82, 10.3233/jad-190367.
- Nir Grossman; David C Bono; Nina Dedic; Suhasa Bangalore Kodandaramaiah; Andrii Rudenko; Ho-Jun Suk; Antonino M. Cassara; Esra Neufeld; Niels Kuster; Li-Huei Tsai; et al.Alvaro Pascual-LeoneEdward S. Boyden Noninvasive Deep Brain Stimulation via Temporally Interfering Electric Fields. Cell 2017, 169, 1029-1041.e16, 10.1016/j.cell.2017.05.024.
- Sangjun Lee; ChanY Lee; Jimin Park; Chang-Hwan Im; Individually customized transcranial temporal interference stimulation for focused modulation of deep brain structures: a simulation study with different head models. Scientific Reports 2020, 10, 1-11, 10.1038/s41598-020-68660-5.
- Linda L. Chao; Effects of Home Photobiomodulation Treatments on Cognitive and Behavioral Function, Cerebral Perfusion, and Resting-State Functional Connectivity in Patients with Dementia: A Pilot Trial. Photobiomodulation, Photomedicine, and Laser Surgery 2019, 37, 133-141, 10.1089/photob.2018.4555.
- Katsuyuki Machii; Daniel Cohen; Ciro Ramos-Estebanez; Alvaro Pascual-Leone; Safety of rTMS to non-motor cortical areas in healthy participants and patients. Clinical Neurophysiology 2006, 117, 455-471, 10.1016/j.clinph.2005.10.014.
- Kamalini G Ranasinghe; Jungho Cha; Leonardo Iaccarino; Leighton B. Hinkley; Alexander J. Beagle; Julie Pham; William J. Jagust; Bruce L. Miller; Katherine P. Rankin; Gil D. Rabinovici; et al.Keith A. VosselSrikantan S. Nagarajan Neurophysiological signatures in Alzheimer’s disease are distinctly associated with TAU, amyloid-β accumulation, and cognitive decline. Science Translational Medicine 2020, 12, eaaz4069, 10.1126/scitranslmed.aaz4069.
- Amy M Pooler; Emma C Phillips; Dawn H W Lau; Wendy Noble; Diane P Hanger; Physiological release of endogenous tau is stimulated by neuronal activity. EMBO reports 2013, 14, 389-394, 10.1038/embor.2013.15.
- Jessica W. Wu; Syed A. Hussaini; Isle M. Bastille; Gustavo A. Rodriguez; Ana Mrejeru; Kelly Rilett; David W. Sanders; Casey Cook; Hongjun Fu; Rick A.C.M. Boonen; et al.Mathieu HermanEden NahmaniSheina EmraniY. Helen FigueroaDavid W Sanders Marc I DiamondCatherine L. ClellandSelina WrayJessica W Wu S Abid Hussaini Isle M Bastille Gustavo A Rodriguez Kelly Rilett Hongjun Fu Rick A C M Boonen Mathieu Herman Eden Nahmani Sheina Emrani Y Helen Figueroa Catherine L Clelland Karen E Duff Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience 2016, 19, 1085-1092, 10.1038/nn.4328.
- Benjamin Cretin; Pharmacotherapeutic strategies for treating epilepsy in patients with Alzheimer’s disease. Expert Opinion on Pharmacotherapy 2018, 19, 1201-1209, 10.1080/14656566.2018.1496237.
- Pascal E. Sanchez; Lei Zhu; Laure Verret; Keith A. Vossel; Anna G. Orr; John R. Cirrito; Nino Devidze; Kaitlyn Ho; Gui-Qiu Yu; Jorge J. Palop; et al.Lennart Mucke Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proceedings of the National Academy of Sciences 2012, 109, E2895-E2903, 10.1073/pnas.1121081109.
- Leslie M Tong; Helen Fong; Yadong Huang; Stem cell therapy for Alzheimer’s disease and related disorders: current status and future perspectives. Experimental & Molecular Medicine 2015, 47, e151-e151, 10.1038/emm.2014.124.
- Jill K. Morris; Jeffrey Burns; Insulin: An Emerging Treatment for Alzheimer’s Disease Dementia?. Current Neurology and Neuroscience Reports 2012, 12, 520-527, 10.1007/s11910-012-0297-0.
- Amy M Pooler; Emma C Phillips; Dawn H W Lau; Wendy Noble; Diane P Hanger; Physiological release of endogenous tau is stimulated by neuronal activity. EMBO reports 2013, 14, 389-394, 10.1038/embor.2013.15.
- Jessica W. Wu; Syed A. Hussaini; Isle M. Bastille; Gustavo A. Rodriguez; Ana Mrejeru; Kelly Rilett; David W. Sanders; Casey Cook; Hongjun Fu; Rick A.C.M. Boonen; et al.Mathieu HermanEden NahmaniSheina EmraniY. Helen FigueroaDavid W Sanders Marc I DiamondCatherine L. ClellandSelina WrayJessica W Wu S Abid Hussaini Isle M Bastille Gustavo A Rodriguez Kelly Rilett Hongjun Fu Rick A C M Boonen Mathieu Herman Eden Nahmani Sheina Emrani Y Helen Figueroa Catherine L Clelland Karen E Duff Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience 2016, 19, 1085-1092, 10.1038/nn.4328.
- Benjamin Cretin; Pharmacotherapeutic strategies for treating epilepsy in patients with Alzheimer’s disease. Expert Opinion on Pharmacotherapy 2018, 19, 1201-1209, 10.1080/14656566.2018.1496237.
- Pascal E. Sanchez; Lei Zhu; Laure Verret; Keith A. Vossel; Anna G. Orr; John R. Cirrito; Nino Devidze; Kaitlyn Ho; Gui-Qiu Yu; Jorge J. Palop; et al.Lennart Mucke Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proceedings of the National Academy of Sciences 2012, 109, E2895-E2903, 10.1073/pnas.1121081109.
- Leslie M Tong; Helen Fong; Yadong Huang; Stem cell therapy for Alzheimer’s disease and related disorders: current status and future perspectives. Experimental & Molecular Medicine 2015, 47, e151-e151, 10.1038/emm.2014.124.
- Jill K. Morris; Jeffrey Burns; Insulin: An Emerging Treatment for Alzheimer’s Disease Dementia?. Current Neurology and Neuroscience Reports 2012, 12, 520-527, 10.1007/s11910-012-0297-0.