Oxidative stress (OS) is mediated by reactive oxygen species (ROS), which in cardiovascular and other disease states, damage DNA, lipids, proteins, other cellular and extra-cellular components. OS is both initiated by, and triggers inflammation, cardiomyocyte apoptosis, matrix remodeling, myocardial fibrosis, and neurohumoral activation. These have been linked to the development of heart failure (HF).
- oxidative stress
- novel markers
- heart failure
1. Overview of ROS and RNS
Heart failure (HF) is a global health problem, with an estimated global prevalence of 64.3 million people in 2017 [1]. Although mortality from cardiovascular diseases (CVD) has declined, the prevalence of HF continues to increase. In the United States alone, the prevalence of HF from 2015–2018 in those over 20 years of age was projected to increase by 3% from 6 to 8 million by 2030 [2,3][2][3]. In the UK, HF increased by 23% from 2002 to 2014, affecting 1.4% of the population [4]. In Southeast Asia, HF was higher at an estimated 4.5% to 6.7% [5]. This increase in HF prevalence is due to an ageing population, improved survival after myocardial infarction, poor adherence to HF prevention strategies, and increasing prevalence of cardiovascular risk factors [6]. The global burden of HF will continue to rise, leading to escalating healthcare expenditure. In 2012, the overall economic cost associated with HF globally was estimated to be USD 108 billion per annum, with most costs related to inpatient hospitalization [7].
2. Normal Physiology of ROS and RNS
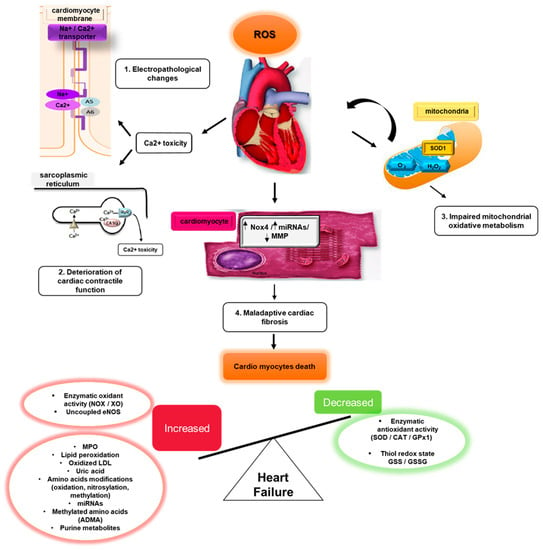
3. ROS/RNS and the Induction of Cardiomyopathy
4. ROS/RNS and Cardiac Fibrosis
5. Oxidative Stress in the Right Ventricle
References
- Bragazzi, N.L.; Zhong, W.; Shu, J.; Abu Much, A.; Lotan, D.; Grupper, A.; Younis, A.; Dai, H. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur. J. Prev. Cardiol. 2021, 28, 1682–1690.
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743.
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States. Circ. Heart Fail. 2013, 6, 606–619.
- GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27.
- Sajedinejad, S.; Majdzadeh, R.; Vedadhir, A.; Tabatabaei, M.G.; Mohammad, K. Maternal mortality: A cross-sectional study in global health. Glob. Health 2015, 11, 4.
- Bahit, M.C.; Kochar, A.; Granger, C.B. Post-Myocardial Infarction Heart Failure. JACC Heart Fail. 2018, 6, 179–186.
- Cook, C.; Cole, G.; Asaria, P.; Jabbour, R.; Francis, D.P. The annual global economic burden of heart failure. Int. J. Cardiol. 2014, 171, 368–376.
- de Jong, J.W.; Schoemaker, R.G.; de Jonge, R.; Bernocchi, P.; Keijzer, E.; Harrison, R.; Sharma, H.S.; Ceconi, C. Enhanced Expression and Activity of Xanthine Oxidoreductase in the Failing Heart. J. Mol. Cell. Cardiol. 2000, 32, 2083–2089.
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570.
- Yasunari, K.; Maeda, K.; Watanabe, T.; Nakamura, M.; Yoshikawa, J.; Asada, A. Comparative effects of valsartan versus amlodipine on left ventricular mass and reactive oxygen species formation by monocytes in hypertensive patients with left ventricular hypertrophy. J. Am. Coll. Cardiol. 2004, 43, 2116–2123.
- Hirotani, S.; Otsu, K.; Nishida, K.; Higuchi, Y.; Morita, T.; Nakayama, H.; Yamaguchi, O.; Mano, T.; Matsumura, Y.; Ueno, H.; et al. Involvement of Nuclear Factor-κB and Apoptosis Signal-Regulating Kinase 1 in G-Protein–Coupled Receptor Agonist–Induced Cardiomyocyte Hypertrophy. Circulation 2002, 105, 509–515.
- Kim, H.E.; Dalal, S.S.; Young, E.; Legato, M.J.; Weisfeldt, M.L.; D'Armiento, J. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J. Clin. Investig. 2000, 106, 857–866.
- Troughton, R.W.; Frampton, C.M.; Brunner-La Rocca, H.P.; Pfisterer, M.; Eurlings, L.W.; Erntell, H.; Persson, H.; O'Connor, C.M.; Moert, D.; Karlström, P. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: An individual patient meta-analysis. Eur. Heart J. 2014, 35, 1559–1567.
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc. Res. 2000, 46, 214–224.
- Spinale, F.G.; Coker, M.L.; Heung, L.J.; Bond, B.R.; Gunasinghe, H.R.; Etoh, T.; Goldberg, A.T.; Zellner, J.L.; Crumbley, A.J. A Matrix Metalloproteinase Induction/Activation System Exists in the Human Left Ventricular Myocardium and Is Upregulated in Heart Failure. Circulation 2000, 102, 1944–1949.
- Spinale, F.G. Matrix metalloproteinases: Regulation and dysregulation in the failing heart. Circ. Res. 2002, 90, 520–530.
- Zhang, H.; Fan, L.; Liao, H.; Tu, L.; Zhang, J.; Xu, D.; Feng, J. Correlations of cardiac function with inflammation, oxidative stress and anemia in patients with uremia. Exp. Ther. Med. 2021, 21, 250.
- Sundström, J.; Evans, J.C.; Benjamin, E.J.; Levy, D.; Larson, M.G.; Sawyer, D.B.; Siwik, D.A.; Colucci, W.S.; Sutherland, P.; Wilson, P.; et al. Relations of Plasma Matrix Metalloproteinase-9 to Clinical Cardiovascular Risk Factors and Echocardiographic Left Ventricular Measures: The Framingham Heart Study. Circulation 2004, 109, 2850–2856.
- Shults, N.V.; Melnyk, O.; Suzuki, D.I.; Suzuki, Y.J. Redox Biology of Right-Sided Heart Failure. Antioxidants 2018, 7, 106.
- Mikhael, M.; Makar, C.; Wissa, A.; Le, T.; Eghbali, M.; Umar, S. Oxidative Stress and Its Implications in the Right Ventricular Remodeling Secondary to Pulmonary Hypertension. Front. Physiol. 2019, 10, 1233.
- DeMarco, V.G.; Whaley-Connell, A.T.; Sowers, J.R.; Habibi, J.; Dellsperger, K.C. Contribution of oxidative stress to pulmonary arterial hypertension. World J. Cardiol. 2010, 2, 316–324.
- Khoo, N.K.; Cantu-Medellin, N.; Devlin, J.E.; St Croix, C.M.; Watkins, S.C.; Fleming, A.M.; Champion, H.C.; Mason, R.P.; Freeman, B.A.; Kelley, E.E. Obesity-induced tissue free radical generation: An in vivo immuno-spin trapping study. Free Radic. Biol. Med. 2012, 52, 2312–2319.
- Zelko, I.N.; Zhu, J.; Roman, J. Role of SOD3 in silica-related lung fibrosis and pulmonary vascular remodeling. Respir. Res. 2018, 19, 221.