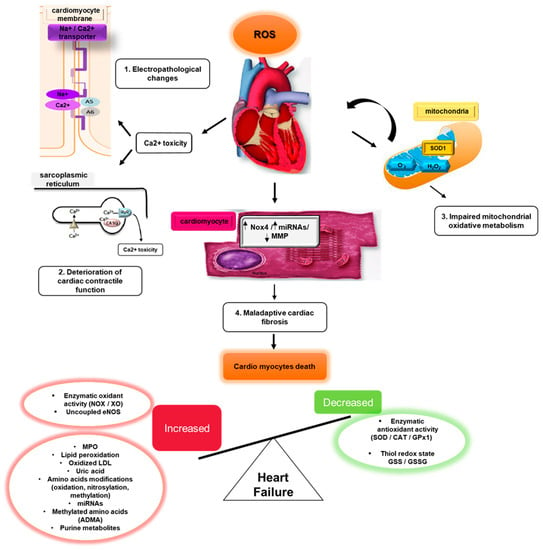
Oxidative stress (OS) is mediated by reactive oxygen species (ROS), which in cardiovascular and other disease states, damage DNA, lipids, proteins, other cellular and extra-cellular components. OS is both initiated by, and triggers inflammation, cardiomyocyte apoptosis, matrix remodeling, myocardial fibrosis, and neurohumoral activation. These have been linked to the development of heart failure (HF).
1. Overview of ROS and RNS
Heart failure (HF) is a global health problem, with an estimated global prevalence of 64.3 million people in 2017 [1]. Although mortality from cardiovascular diseases (CVD) has declined, the prevalence of HF continues to increase. In the United States alone, the prevalence of HF from 2015–2018 in those over 20 years of age was projected to increase by 3% from 6 to 8 million by 2030 [2][3][2,3]. In the UK, HF increased by 23% from 2002 to 2014, affecting 1.4% of the population [4]. In Southeast Asia, HF was higher at an estimated 4.5% to 6.7% [5]. This increase in HF prevalence is due to an ageing population, improved survival after myocardial infarction, poor adherence to HF prevention strategies, and increasing prevalence of cardiovascular risk factors [6]. The global burden of HF will continue to rise, leading to escalating healthcare expenditure. In 2012, the overall economic cost associated with HF globally was estimated to be USD 108 billion per annum, with most costs related to inpatient hospitalization [7].
OS is caused by excessive production of Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS), two major redox biomarkers. Sies first defined it as a disturbance in the prooxidant-antioxidant balance in 1985
[8][32]. ROS act as intermediates in cellular pathways and can be classified into either free radicals such as superoxide (O
2−) or hydroxyl (·OH) or non-free radicals such as hydrogen peroxide (H
2O
2) and peroxynitrite (ONOO
−). Reactive nitrogen species (RNS) are species derived from nitric oxide and superoxide through the enzymatic activity of inducible nitric oxide synthase 2 (NOS
2) and NOX
[8][32].
2. Normal Physiology of ROS and RNS
Normally, ROS/RNS are generated in small amounts in the heart by enzymes such as NADPH oxidases, nitric oxide synthase (NOS), xanthine oxidase (XO) and mitochondria and are play a pivotal role in modulating cell cycle homeostasis, excitation–contraction coupling and cell function. NAPDH oxidases are transmembrane enzymes that specialize in producing O
2− from molecular oxygen by transfer of an unpaired electron from NADPH. The major isoform of NOX in the heart is NOX4, which is expressed primarily in the mitochondria of cardiac myocytes
[9][33]. Inhibition of XO with allopurinol or oxypurinol in animal HF models led to protection of myocardial contractility, improved left ventricular function and the reversal of maladaptive cardiac remodeling
[8][32].
NOS is responsible for endogenous production of nitric oxide (NO), an important signaling molecule in humans. When tetrahydrobiopterin (BH4) is sufficient, L-arginine and O2 are catalyzed by NOS to become l-citrulline and NO. However, in pathological conditions, uncoupling of NOS occurs and O2− is generated as a by-product of NO synthesis during electron leakage at the electron transport chains during mitochondrial respiration. Complexes I and III of the electron transport chain are major sites of ROS generation, but under pathological conditions, the ROS from mitochondria is markedly increased.
3. ROS/RNS and the Induction of Cardiomyopathy
Cardiac hypertrophy and maladaptive remodeling are common in patients with HF, with progressive cardiomyocyte hypertrophy contributing to the development of HF. ROS/RNS is implicated in the downstream mechanistic underpinning of cardiomyocyte hypertrophy due to RAAS and sympathetic nervous system activation. Yasunari et al. demonstrated a significant correlation between left ventricular mass index and ROS/RNS levels in 104 hypertensive patients with left ventricular hypertrophy
[10][34]. Hirotani et al. added that ROS/RNS is involved in the activation of NF-κB by G-protein-coupled receptor agonists such as Ang II or ET-1
[11][35]. Animal studies have also shown a close link between ROS and cardiomyocyte hypertrophy, which can be slowed by antioxidants.
An important pathway, Ang II induces cardiac hypertrophy by activating angiotensin II (AT1R) receptor, which induces ROS/RNS generation and activation of multiple downstream signaling kinases. This promotes cardiomyocyte survival and suppression of apoptosis. The c-Rel subunit of NF-κB has also been identified as a major promoter of cardiac hypertrophy in mice. Ang II stimulation of the Nox4-histone deacetylase (HDAC) axis leads to increased ROS production and nuclear export of HDAC. HDAC suppresses pro-hypertrophic transcription factors, but with HDAC loss, this suppressive effect is lost.
4. ROS/RNS and Cardiac Fibrosis
Cardiac fibrosis is a major pathological mechanism in HF (Figure 1), with increased extracellular matrix deposition from cardiac fibroblasts transitioning to secretory myofibroblasts. ROS and RNS mediate cardiac fibrosis, and Ang II upregulates transforming growth factor-β1 (TGF-β1) expression by activating AT1R and NOX4.
Figure 1. Mechanisms and expression of OS biomarkers in heart failure. Excessive ROS/RNS leads to cardiomyocyte hypertrophy and cardiac fibrosis in heart failure.
Matrix metalloproteinases (MMPs) are known to be pathologically activated by ROS through binding and opening of active sites on MMPs. This prevents TIMPs (Tissue Inhibitors of MMPs) from accessing the active site
[12][36], ultimately ceasing activity and causing the onset of cardiac fibrosis. MMPs also regulate extracellular matrix remodeling, and alteration can lead to abnormal deposition and eventually dilated cardiomyopathy
[13][30]. Left ventricular remodeling and HF onset is also thought to have been caused by MMPs
[14][15][16][37,38,39], and have been associated with increased left ventricular internal dimensions and wall thickness
[17][40]. This suggests that MMP may be a mediator and possible marker of cardiac remodeling and can be regulated by various ROS-related pathways, such as Ang-II induction of NF-κB and AP-1 transcription factors
[18][41]. Aldosterone, part of the renin–angiotensin–aldosterone cascade (RAAS) commonly activated in HF, has altered levels of MMPs in animal studies. ROS/RNS has also been shown to affect MMPs directly by protein modification. These findings have been validated in several in vivo and in vitro studies
[14][15][16][37,38,39].
5. Oxidative Stress in the Right Ventricle
The right ventricle (RV) is highly susceptible to OS compared to the left ventricle (LV) due to its inability to regulate expression of manganese superoxide dismutase, a key enzyme that attenuates ROS
[19][42]. A high degree of OS adversely affects pulmonary vasculature and induces RV remodeling
[20][43]. Pulmonary vascular remodeling due to OS may lead to hypertrophy and HF.
A key condition of OS-mediated HF is pulmonary hypertension (PH). During PH development, ROS generation increases due to monocytes accumulating in pulmonary arterioles
[21][44]. An animal experimental model by Khoo et al.
[22][45] revealed high radical formation in high-fat diets of rats. Investigating the importance of the balance between antioxidants and oxidants, Zelko et al.
[23][46] knocked out SOD3 in mice and induced PH using silica. The resulting mice exhibited much higher RV pressures compared to the wildtype.