Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Ilyas Chachoua.
Secondary acute myeloid leukemia (sAML) is a heterogeneous malignant hematopoietic disease that arises either from an antecedent hematologic disorder (AHD) including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), aplastic anemia (AA), or as a result of exposure to genotoxic chemotherapeutic agents or radiotherapy (therapy related AML, tAML). sAML is diagnosed when the number of blasts is ≥20% in the bone marrow or peripheral blood, and it is characterized by poor prognosis, resistance to therapy and low overall survival rate.
- sAML
- AHD
- MPN
- MDS
- tAML
- 7 + 3 regimen
- AlloSCT
- CAR-T
- HSC
- LSC
1. Introduction
Acute myeloid leukemia (AML) is defined as a heterogeneous malignant clonal disorder of hematopoietic stem cells (HSC) and is the most common myeloid disorder among adults. This disease can be secondary (sAML) to either an AHD such as MPN and MDS or as a consequence of a prior treatment (tAML), or without an AHD history in the case of de novo AML [1,2,3][1][2][3]. MPNs lack cytopenia and are instead characterized by heightened differentiation of progenitor cells and are negative for the BCR-ABL fusion protein. They are divided into three main sub-categories: polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) [4]. MPN rates of progression to sAML vary by subtype: on average 15% of PMF patients, 8.35% of PV patients, and 1.85% of ET patients develop sAML over a ten-year period [5]. MDS are narrow clonal stem cell disorders characterized by heightened cytopenia in the blood and bone marrow due to apoptosis of hematopoietic progenitor cells, and one third of these syndromes progress to sAML [6,7][6][7]. AA is a rare, life-threatening bone marrow disorder characterized by deficiencies in hematopoietic cell production resulting from T-cell mediated autoimmunity. Like other AHDs, approximately 15–20% of AA patients over a ten-year period progress further to MDS/sAML. In addition to mutations and chromosomal abnormalities (Figure 1), other factors including telomere attrition, time to therapy, and the patient response to initial immunosuppressant treatment contribute to disease progression [8,9][8][9].
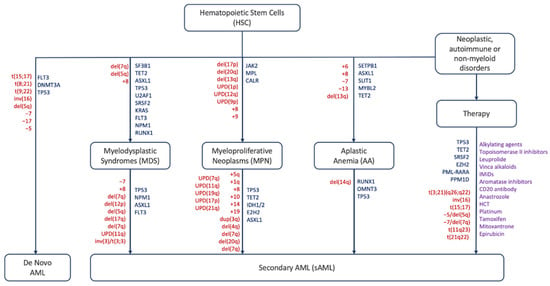
Figure 1. Schematic representation of mutations, abnormalities and factors driving different hematological disorders and their progression to AML/sAML. Red: chromosomal abnormalities, blue: gene mutations, purple: therapeutic agents [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26].
Unlike AHDs, sAML is a severe disease with a poor prognosis that has an overall survival time of 4.7 months and an event-free survival time of 2.9 months [27]. The disease affects the elderly and the majority of diagnosed cases are over 65 years old: the median age of diagnosis is 67 years old with a third of patients over the age of 75. Common mutations that lead to the evolution of sAML are found in members of the spliceosome such as SRSF2, epigenetic modifiers including TET2, IDH1/2, ASXL1, and EZH2, or TP53 which maintains genomic integrity [28]. The aforementioned mutations are acquired on top of the mutations driving MDS or MPN development [29,30,31,32,33,34,35][29][30][31][32][33][34][35].
According to the 2016 WHO classification, patients are diagnosed with sAML when the percentage of myeloblasts in the bone marrow and/or peripheral blood is equal to or greater than 20% [36,37][36][37]. Although blast count has been set as the differentiator between the three phases, there are limitations to this method in that the blasts under examination, commonly through microscopy, are not easily distinguishable between normal samples and those of MDS and MPN patients. In addition to blast count, there are other indicators of progression to sAML, such as decreased apoptosis in the case of post-MDS sAML and increased cell proliferation for post-MPN sAML [38,39,40][38][39][40]. The vast majority of sAML patients progress from MDS (~85%) and upon exposure to therapy (~9%) (Figure 2). MPN and AA contribution is lower with (~5%) and (~1%), respectively [5,7,41,42,43,44][5][7][41][42][43][44].
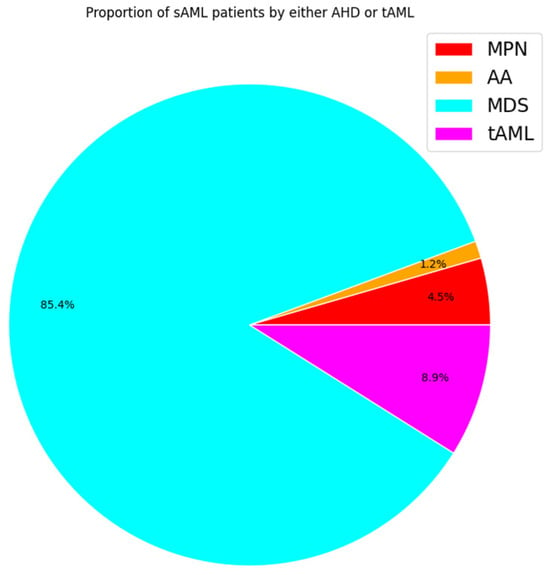
Figure 2. Pie chart displaying estimated proportions of sAML patients by history based on studies monitoring leukemic transformation. Each color represents the proportion of sAML patients by class. Percentages based on average incidence per 100,000 people.
The outcome of sAML patients correlates with the mutational landscape. For example, patients with TP53 mutations, roughly 10–15%, have a worse outcome than those with wild type TP53. Not just due to the pernicious nature of these mutations, but also because of the co-occurring mutations including but not limited to IDH2 and NPM1. Due to the difference in mutation profiles between sAML and de novo AML, the treatment regimen has not been as successful when applied to sAML. For example, TP53 mutant patients had an overall survival of 8 months with induction therapy compared with 1.7 months for those without. The hazard ratio for having a TP53 mutation was 3.1-fold which is higher than either increasing age or performance status, although co-occurring mutations in FLT3 had a slightly smaller ratio of 3.01 [45]. The most recommended method for treating sAML is allogeneic stem cell transplantation (alloSCT) due to the highest probability of success [46], especially compared with the more traditional method of 7 + 3, or 7 days of continuous dosing of cytarabine followed by 3 days of IV injection of daunorubicin, which has been the standard for decades [47]. However, direct comparison studies have shown that sAML patients are consistently less responsive to 7 + 3 treatment compared with de novo AML, and have a lower overall survival rate with this regimen, prompting the need for new therapies to be developed [48,49][48][49].
2. Pathophysiology of sAML
The WHO standard of a ≥20% blast count to differentiate between sAML and antecedent disorders is arbitrary like any threshold, but also has a potentially decisive impact on patients who may either display other characteristics of sAML with a blast count below the threshold or do not display characteristics of leukemogenesis despite having surpassed the threshold [50]. Therefore, this approach does not always assure diagnosis accuracy or reflect the complexity of leukemogenesis nor does it guarantee the optimal treatment for the patient. It is crucial to note that there are additional factors such as mutation type influencing diagnosis and treatment regimens. During the progression process, the immune system responds to the growth of the malignant cells. For example, Bauer et al. have shown a shift in immune cell populations between healthy donors’ bone marrow samples and those diagnosed with either MDS or sAML: there were neither CD3+CD8+ nor CD3+FOXP3+ T cells within a 25-micron radius in healthy bone marrow samples, but both of these populations were present in sAML patients at much higher levels. However, when comparing subsets of sAML, the outcomes were not uniform: in contrast to patients with TP53 mutations, patients with mutations in either signal transduction genes, chromatin modifiers, or splicing factors showed a significant increase in both populations [51]. One of the reasons why sAML is more common in older patients, especially over the age of 60, is the phenomenon of clonal hematopoiesis of indeterminate potential (CHIP), which is defined as having more than 2% mutant hematopoietic stem cells (HSC) without a history of either cytopenia or myeloid neoplasms. The risk for patients accumulates every year by 0.5–1%, thus most sAML patients skew older with some exceptions such as Fanconi anemia (FA) that progresses earlier. FA is a rare blood disorder characterized by a selective growth advantage to HSCs with an extra copy of 1q, leading to bone marrow depletion and an elevated risk of both MDS and later sAML [52,53][52][53]. However, in cases of cytopenia where patients do not exactly meet the criteria for MDS, they are diagnosed with clonal cytopenia of undetermined significance (CCUS) [54]. A 2023 study of UK Biobank patients found that patients diagnosed with either CCUS or CHIP had a significantly higher risk to develop MPNs and subsequently sAML: 1.74 for the former and 2.63 for the latter. For comparison, the risk for patients over 65 to develop MPN/sAML was 1.53 and the risk associated with the total number of mutations was 2.32. Non-genomic factors like red blood cell width distribution (RDW) over 15% or the mean corpuscular volume over 100 fL (MCV) had even higher risks: 3.63 and 4.03 [55]. RDW is also a biomarker for leukemic transformation: a higher RDW is not only used to distinguish MDS patients from healthy ones, but it is also a reliable predictor of leukemogenesis years after the initial MDS diagnosis. Higher RDW is associated with overall worse outcome in patients who have been treated with alloSCT and increased possibility to have passenger mutations in genes like NPM1 or ASXL1 [56]. In terms of cytogenetic risk for leukemic transformation, chromosomal mosaicism is also positively correlated with MPN formation: the 10-year cumulative incidence with mosaicism was 83% and 43% without it [55]. Moreover, chromosomal abnormalities impact diagnosis. For example, t(8;21) is associated with a favorable diagnosis, whereas poor prognosis is associated with −7, inv(3)/t(3q)/del(3q), −7/del(7q), or complex karyotype (CK) with ≥3 abnormalities, which substantially increases the risk of leukemic progression [57,58][57][58]. A recent study has reported a case of a 44-year-old female with MDS/MPN where constitutional trisomy 21 was the only identified chromosomal abnormality [59]. Due to higher average age, the risk of an adverse karyotype is higher in sAML than in de novo. Compared with de novo AML, sAML patients have lower overall platelet and leukocyte counts, as well as a lower blast percentage in either bone marrow or peripheral blood [60]. Another study found that 81% of post-MDS sAML patients had a lower WBC count compared with 68% of MPN blast phase or 60% of de novo AML cases (Table 1) [61].Table 1.
Hematological features associated with MPN, MDS, sAML and de novo AML.
| Clinical Overview | Healthy | MDS | MPN | sAML | De Novo AML |
|---|
Table 2.
Mutated genes implicated in leukemic transformation and clonal expansion.
| Category of Genes | Examples | Citations | |||
|---|---|---|---|---|---|
| Age (y) | 18–65 | 53–98 | 18–92 | 21–77 | 18–59 |
| Spliceosome | SRSF2, U2AF1, SF3B1 | [86,87][83] | |||
| White Blood Cells (109/L) | 4–11 | 1.1–17.9 | 7.2–14.7 | 0.8–144.1 | 0.77–419.9 |
| Platelets (109/L) | 150–450 | 8–505 | 376–720 | 3–752 | 30–171 |
| Hemoglobin (g/L) | 120–175 | 47–149 | 109–173 | 34–143 | 2–1726 |
| Clinical record | Free of treatment and transfusion | Both therapy free and treatment * | 3 + 7 regimen Hypomethylation or palliative treatment |
3 + 7 regimen | |
| Reference | [62] | [63] | [64] | [65] | [65] |
* anti-platelet drugs, cytoreductive therapy, JAK-2 inhibitors, immunomodulators, venipuncture, and supportive RBC transfusions.
Another factor that contributes to sAML progression is inflammation through immune system dysregulation. Patients with autoimmune diseases (AIDs) are already at higher risk of developing sAML due to the elevated levels of inflammatory cytokines in the blood [66,67][66][67]. A recent study demonstrated that the increase in inflammation is particularly observed in MPN patients with TP53 mutations, either heterozygous or multi-hit, where the mutant myeloid cells gain a selective advantage over erythroid cells, especially those with WT TP53, which leads to a distortion in the ratio between erythroid and myeloid progenitor cells [68]. For example, TNF drives malignant clonal dominance by targeting healthy myeloid progenitor cells with both apoptotic and necroptotic signaling while malignant cells are left unaffected and able to proliferate through immuno-evasive mechanisms. Moreover, constitutive NFκB activity has been reported in both MPN/MDS and sAML patients [68,69,70][68][69][70]. In the case of MDS progressing to sAML, the chemokine receptor CCRL2, normally expressed in granulocytes, monocytes and NK cells is up-regulated in stem cells, which in turn stimulates IL-8 and the chemokine receptor CXCR2 [71,72][71][72]. On the other hand, IL-8 is a catalyst for several downstream pathways that promote proliferation, especially in tumors, including NFκB, MAPK, AKT, STAT3, and β-catenin. In a study conducted by Montes et al., compared with healthy donors, patients with MDS and sAML have significantly reduced counts of both CD4+ T lymphocytes and NK cells, with sAML having a higher count of CD4+ T lymphocytes than MDS, but lower than healthy donors. This illustrates that at least the correlation between CD4+ T lymphocytes and MDS progression to sAML is not linear. In tandem with lower counts of proactive immune cells, programmed death ligand 1 (PD-L1) is upregulated, which suppresses the T-cell response to tumor growth and permits clonal expansion and metastasis of leukemic cells [73,74][73][74]. Patients with sAML have lower or similar expression levels of PD-L1 compared with MPN/MDS, with no difference between early and advanced stages of MDS; suggesting that the peak of PD-L1 expression results in a long-term suppression of the immune response that allows subsequent mutations to develop and trigger the progression to sAML [75]. In parallel, monocytic myeloid-derived suppressor cells (Mo-MDSC), another immuno-suppressive cell type, has been shown to have stronger positive correlation with the progression to sAML from MDS [74,76][74][76]. In addition, alteration in the extracellular matrix (ECM), and in particular the leucine-rich proteoglycan biglycan (BG), contributes to the heightened inflammatory environment observed in both MPN/MDS and sAML. BG is expressed in the bone marrow of both MDS and sAML patients but not in healthy individuals: it promotes cell signaling, bone mineralization, and differentiation. The presence of BG was positively correlated with activity of inflammasome components such as IL-1β, IL-18, and IFN-α. There was no significant difference in BG bone marrow expression between MDS and sAML patients. The hazard ratio of BG-high MDS patients versus BG-low patients for progression to sAML was 8.3 [77].
Another key feature of sAML is the increased self-renewal activity of pre- Leukemic Stem Cells (pre- LSC) through the WNT/β-catenin pathway activation during progression, which produces three main LSC phenotypes: multi-potent progenitor (MPP), lymphoid primed multi-potent progenitor (LMPP) and granulocyte-macrophage progenitor (GMP) [78]. Compared with de novo AML, sAML patients had higher amounts of MPP-like LSCs and LMPP-like LSCs, and this difference was more pronounced in post-MPN sAML. Post-MDS sAML patients had more GMP-like LSCs than post-MPN patients, but similar to de novo AML. The first two types of LSCs were strongly correlated with poor prognosis while GMP-like LSCs were more commonly seen in patients (either de novo or sAML) with either an intermediate or favorable prognosis. There was no difference in terms of LSC type distribution between patients younger than 65 and those older than 65 [79].
In contrast to increased pre-LSC activity in MPN patients progressing to sAML, there is a negative correlation between interferon (IFN) activity and risk of sAML. A study by de Castro et al. categorized MPN patients by both LSC and IFN activity and found that those with both the lowest IFN and highest LSC activity had the greatest risk of progression to sAML. Clonogenicity was significantly higher in this cohort compared with the rest of the study population, and the result was the same when comparing before and after transformation. The low IFN activity in these transforming cells also results in a more chaotic microenvironment where endothelial cells are dysregulated, and leukocytes are behaving abnormally while under increased oxidative stress [80].
The transition to sAML is accompanied with a shift in the clonal architecture inside the bone marrow. This shift is correlated with the number of acquired mutations during progression. Static shift occurs when mutations are acquired sequentially and the clones with the most mutations gradually dominate the bone marrow. Dynamic-S (for single nucleotide variant) shift occurs upon acquisition of multiple mutations that can be in multiple categories simultaneously (Table 2) and their rise to clonal dominance is expedited. Finally, the Dynamic-C (for chromosomal) shift is similar to Dynamic-S except that instead of gaining mutations, the clones acquire chromosomal abnormalities that confer a selective advantage (Table 3) [81
| [ | |||||
| 84 | ] | ||||
| Deletions | del(7q), del(5q), del(17p) | [106,113][95][96] | DNA Methylation | DMNT3A, TET 1/2, IDH 1/2, | [88,89][85][86] |
| Duplications | dup(1q), dup(3q), dup(11q), dup(17q) | [114,115,116][97][98][99] | Activated Signaling | CALR, JAK2, PTPN11, TpoR, KRAS, FLT3, NRAS | [90,91] |
| Translocations | [ | 87][88] | |||
| t(1;11)(q21;p15), t(10;11)(q22;q23), t(8;21) | [ | 65,117,118][65][100][101] | Transcription Factors | RUNX1, NFE2, TP53 | [92,,96][8993,][90 |
| Inversions | 94 | ,][91]95[92][93] | |||
| inv(3)/t(3;3) | Chromatin Modification | EZH2, ASXL1, NPM1 | [89,97][86][94] |
References
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907.
- Higgins, A.; Shah, M.V. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes 2020, 11, 749.
- Pedersen-Bjergaard, J.; Andersen, M.K.; Andersen, M.T.; Christiansen, D.H. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008, 22, 240–248.
- McMullin, M.F.; Anderson, L.A. Aetiology of Myeloproliferative Neoplasms. Cancers 2020, 12, 1810.
- Shahin, O.A.; Chifotides, H.T.; Bose, P.; Masarova, L.; Verstovsek, S. Accelerated Phase of Myeloproliferative Neoplasms. Acta Haematol. 2021, 144, 484–499.
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252.
- Dan, C.; Chi, J.; Wang, L. Molecular mechanisms of the progression of myelodysplastic syndrome to secondary acute myeloid leukaemia and implication for therapy. Ann. Med. 2015, 47, 209–217.
- Sun, L.; Babushok, D.V. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood 2020, 136, 36–49.
- Wang, L.; Liu, H. Pathogenesis of aplastic anemia. Hematology 2019, 24, 559–566.
- Cook, M.R.; Karp, J.E.; Lai, C. The spectrum of genetic mutations in myelodysplastic syndrome: Should we update prognostication? eJHaem 2022, 3, 301–313.
- Heuser, M.; Gabdoulline, R.; Löffeld, P.; Dobbernack, V.; Kreimeyer, H.; Pankratz, M.; Flintrop, M.; Liebich, A.; Klesse, S.; Panagiota, V.; et al. Individual outcome prediction for myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia from MDS after allogeneic hematopoietic cell transplantation. Ann. Hematol. 2017, 96, 1361–1372.
- Hussein, K.; Van Dyke, D.L.; Tefferi, A. Conventional cytogenetics in myelofibrosis: Literature review and discussion. Eur. J. Haematol. 2009, 82, 329–338.
- Rubin, C.M.; Larson, R.A.; Anastasi, J.; Winter, J.N.; Thangavelu, M.; Vardiman, J.W.; Rowley, J.D.; Le Beau, M.M. t(3;21)(q26;q22): A recurring chromosomal abnormality in therapy- related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990, 76, 2594–2598.
- Andersen, M.K.; Larson, R.A.; Mauritzson, N.; Schnittger, S.; Jhanwar, S.C.; Pedersen-Bjergaard, J. Balanced chromosome abnormalities inv(16) and t(15;17) in therapy-related myelodysplastic syndromes and acute leukemia: Report from an International Workshop †. Genes Chromosom. Cancer 2002, 33, 395–400.
- Kotsiafti, A.; Giannakas, K.; Christoforou, P.; Liapis, K. Progress toward Better Treatment of Therapy-Related AML. Cancers 2023, 15, 1658.
- Hosono, N. Genetic abnormalities and pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892.
- Cheng, Y.; Wang, Y.; Wang, H.; Chen, Z.; Lou, J.; Xu, H.; Wang, H.; Qian, W.; Meng, H.; Lin, M.; et al. Cytogenetic profile of de novo acute myeloid leukemia: A study based on 1432 patients in a single institution of China. Leukemia 2009, 23, 1801–1806.
- Heuser, M.; Schlarmann, C.; Dobbernack, V.; Panagiota, V.; Wiehlmann, L.; Walter, C.; Beier, F.; Ziegler, P.; Yun, H.; Kade, S.; et al. Genetic characterization of acquired aplastic anemia by targeted sequencing. Haematologica 2014, 99, e165.
- Milosevic, J.D.; Puda, A.; Malcovati, L.; Berg, T.; Hofbauer, M.; Stukalov, A.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, H.; Gisslinger, B.; et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am. J. Hematol. 2012, 87, 1010–1016.
- Maciejewski, J.P.; Risitano, A.; Sloand, E.M.; Nunez, O.; Young, N.S. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood 2002, 99, 3129–3135.
- Strickland, S.A.; Vey, N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2022, 171, 103607.
- Keung, Y.-K.; Pettenati, M.J.; Cruz, J.M.; Powell, B.L.; Woodruff, R.D.; Buss, D.H. Bone Marrow Cytogenetic Abnormalities of Aplastic Anemia. J. Hematol. 2001, 66, 167–171.
- Van Gelder, M.; Schetelig, J.; Volin, L.; Maertens, J.; Socié, G.; Petersen, E.; Thomssen, H.; Biezen, A.; Brand, R.; Witte, T.M.; et al. Monosomal Karyotype Predicts Poor Outcome for MDS/sAML Patients with Chromosome 7 Abnormalities After Allogeneic Stem Cell Transplantation for MDS/sAML. A Study of the MDS Subcommittee of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Blood 2009, 114, 293.
- Birdwell, C.; Fiskus, W.; Kadia, T.M.; Di Nardo, C.D.; Mill, C.P.; Bhalla, K.N. EVI1 dysregulation: Impact on biology and therapy of myeloid malignancies. Blood Cancer J. 2021, 11, 64.
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581.
- Thoennissen, N.H.; Kawamata, N.; Lasho, T.L.; Weiss, T.; Nowak, D.; Kato, M.; Takita, J.; Sanada, M.; Haferlach, T.; Mesa, R.A.; et al. Genomic Changes Associated with Leukemic Transformation of Myeloproliferative Disorders. Blood 2008, 112, 3371.
- Venton, G.; Courtier, F.; Charbonnier, A.; D’incan, E.; Saillard, C.; Mohty, B.; Mozziconacci, M.J.; Birnbaum, D.; Murati, A.; Vey, N.; et al. Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am. J. Hematol. 2018, 93, 330–338.
- Dunbar, A.J.; Rampal, R.K.; Levine, R. Leukemia secondary to myeloproliferative neoplasms. Blood 2020, 136, 61–70.
- Schwind, S.; Jentzsch, M.; Kubasch, A.S.; Metzeler, K.H.; Platzbecker, U. Myelodysplastic syndromes: Biological and therapeutic consequences of the evolving molecular aberrations landscape. Neoplasia 2021, 23, 1101–1109.
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061.
- James, C.; Ugo, V.; Casadevall, N.; Constantinescu, S.N.; Vainchenker, W. A JAK2 mutation in myeloproliferative disorders: Pathogenesis and therapeutic and scientific prospects. Trends Mol. Med. 2005, 11, 546–554.
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476.
- Pikman, Y.; Lee, B.H.; Mercher, T.; Mcdowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270.
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405.
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Mesa, R.A.; Verstovsek, S.; Cervantes, F.; Barosi, G.; Reilly, J.T.; Dupriez, B.; Levine, R.; Le Bousse-Kerdiles, M.C.; Wadleigh, M.; Campbell, P.J.; et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and. Leuk. Res. 2007, 31, 737–740.
- Jilg, S.; Reidel, V.; Müller-Thomas, C.; König, J.; Schauwecker, J.; Höckendorf, U.; Huberle, C.; Gorka, O.; Schmidt, B.; Burgkart, R.; et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia 2016, 30, 112–123.
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Mill, C.P.; Qian, Y.; Raina, K.; Rajapakshe, K.; Coarfa, C.; Soldi, R.; Bose, P.; et al. Targeting nuclear β-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia 2018, 33, 1373–1386.
- Parker, J.E.; Mufti, G.J.; Rasool, F.; Mijovic, A.; Devereux, S.; Pagliuca, A. The role of apoptosis, proliferation, and the Bcl-2–related proteins in the myelodysplastic syndromes and acute myeloid leukemia secondary to MDS. Blood 2000, 96, 3932–3938.
- Cogle, C.R. Incidence and Burden of the Myelodysplastic Syndromes. Curr. Hematol. Malig. Rep. 2015, 10, 272.
- Vaht, K.; Göransson, M.; Carlson, K.; Isaksson, C.; Lenhoff, S.; Sandstedt, A.; Uggla, B.; Winiarski, J.; Ljungman, P.; Brune, M.; et al. Incidence and outcome of acquired aplastic anemia: Real-world data from patients diagnosed in Sweden from 2000–2011. Haematologica 2017, 102, 1683.
- Prébet, T.; Gore, S.D.; Thépot, S.; Esterni, B.; Quesnel, B.; Beyne Rauzy, O.; Dreyfus, F.; Gardin, C.; Fenaux, P.; Vey, N. Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br. J. Haematol. 2012, 157, 764.
- Nilsson, C.; Linde, F.; Hulegårdh, E.; Garelius, H.; Lazarevic, V.; Antunovic, P.; Cammenga, J.; Deneberg, S.; Eriksson, A.; Jädersten, M.; et al. Characterization of therapy-related acute myeloid leukemia: Increasing incidence and prognostic implications. Haematologica 2023, 108, 1015.
- Yanada, M.; Yamamoto, Y.; Iba, S.; Okamoto, A.; Inaguma, Y.; Tokuda, M.; Morishima, S.; Kanie, T.; Mizuta, S.; Akatsuka, Y.; et al. TP53 mutations in older adults with acute myeloid leukemia. Int. J. Hematol. 2016, 103, 429–435.
- Michelis, F.V.; Atenafu, E.G.; Gupta, V.; Kim, D.D.; Kuruvilla, J.; Lipton, J.H.; Loach, D.; Seftel, M.D.; Uhm, J.; Alam, N.; et al. Comparable outcomes post allogeneic hematopoietic cell transplant for patients with de novo or secondary acute myeloid leukemia in first remission. Bone Marrow Transplant. 2015, 50, 907–913.
- Rowe, J.M. The “7 + 3” regimen in acute myeloid leukemia. Haematologica 2022, 107, 3.
- Hulegårdh, E.; Nilsson, C.; Lazarevic, V.; Garelius, H.; Antunovic, P.; Rangert Derolf, Å.; Möllgård, L.; Uggla, B.; Wennström, L.; Wahlin, A.; et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: A report from the Swedish Acute Leukemia Registry. Am. J. Hematol. 2015, 90, 208–214.
- Østgård, L.S.G.; Medeiros, B.C.; Sengeløv, H.; Nørgaard, M.; Andersen, M.K.; Dufva, I.; Friis, L.S.; Kjeldsen, E.; Marcher, C.W.; Preiss, B.; et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: A national population-based cohort study. J. Clin. Oncol. 2015, 33, 3641–3649.
- Estey, E.; Hasserjian, R.P.; Döhner, H. Distinguishing AML from MDS: A fixed blast percentage may no longer be optimal. Blood 2022, 139, 323–332.
- Bauer, M.; Vaxevanis, C.; Al-Ali, K.; Jaekel, N.; Le, C.; Naumann, H.; Schaffrath, J.; Rau, A.; Seliger, B.; Wickenhauser, C. Altered Spatial Composition of the Immune Cell Repertoire in Association to CD34 + Blasts in Myelodysplastic Syndromes and Secondary Acute Myeloid Leukemia. Cancers 2021, 13, 186.
- Hasserjian, R.P.; Steensma, D.P.; Graubert, T.A.; Ebert, B.L. Clonal hematopoiesis and measurable residual disease assessment in acute myeloid leukemia. Blood 2020, 135, 1729–1738.
- Sebert, M.; Phanie Gachet, S.; Leblanc, T.; Bluteau, D.; Gis Peffault De Latour, R.; Correspondence, J.S. Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in Fanconi anemia. Cell Stem Cell 2023, 30, 153–170.e9.
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378.
- Weeks, L.D.; Niroula, A.; Neuberg, D.; Wong, W.; Lindsley, R.C.; Luskin, M.R.; Berliner, N.; Stone, R.M.; DeAngelo, D.J.; Soiffer, R.J.; et al. Prediction of Risk for Myeloid Malignancy in Clonal Hematopoiesis. NEJM Evid. 2023, 2, EVIDoa2200310.
- Vucinic, V.; Ruhnke, L.; Sockel, K.; Rohnert, M.A.; Backhaus, D.; Brauer, D.; Franke, G.N.; Niederwieser, D.; Bornhauser, M.; Rollig, C.; et al. The diagnostic red blood cell distribution width as a prognostic factor in acute myeloid leukemia. Blood Adv. 2021, 5, 5584–5587.
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465.
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J. Clin. Oncol. 2012, 30, 820–829.
- O’Hagan Henderson, S.; Glaser, A.; Frietsch, J.J.; Hochhaus, A.; Hilgendorf, I. The incidental discovery of a constitutional trisomy 21 mosaicism in an adult female with myelodysplastic/myeloproliferative neoplasm. Ann. Hematol. 2022, 101, 919.
- Begna, K.H.; Ali, W.; Naseema Gangat Elliott, M.A.; Al-Kali, A.; Litzow, M.R.; Christopher Hook, C.; Wolanskyj-Spinner, A.P.; Hogan, W.J.; Patnaik, M.M.; Pardanani, A.; et al. Mayo Clinic experience with 1123 adults with acute myeloid leukemia. Blood Cancer J. 2021, 113, 1–8.
- Gupta, V.; Kim, S.; Hu, Z.H.; Liu, Y.; Aljurf, M.; Bacher, U.; Beitinjaneh, A.; Cahn, J.Y.; Cerny, J.; Copelan, E.; et al. Comparison of outcomes of HCT in blast phase of BCR-ABL1− MPN with de novo AML and with AML following MDS. Blood Adv. 2020, 4, 4748–4757.
- Ware, A.D. The complete blood count and white blood cell differential. In Contemporary Practice in Clinical Chemistry; Academic Press: Cambridge, MA, USA, 2020; pp. 429–444.
- Boutault, R.; Peterlin, P.; Boubaya, M.; Sockel, K.; Chevallier, P.; Garnier, A.; Guillaume, T.; Le Bourgeois, A.; Debord, C.; Godon, C.; et al. A novel complete blood count-based score to screen for myelodysplastic syndrome in cytopenic patients. Br. J. Haematol. 2018, 183, 736–746.
- Horvat, I.; Boban, A.; Zadro, R.; Antolic, M.R.; Serventi-Seiwerth, R.; Roncevic, P.; Radman, I.; Sertic, D.; Vodanovic, M.; Pulanic, D.; et al. Influence of Blood Count, Cardiovascular Risks, Inherited Thrombophilia, and JAK2 V617F Burden Allele on Type of Thrombosis in Patients with Philadelphia Chromosome Negative Myeloproliferative Neoplasms. Clin. Lymphoma Myeloma Leuk. 2019, 19, 53–63.
- Wang, S.Y.; Cheng, W.Y.; Mao, Y.F.; Zhu, Y.M.; Liu, F.J.; Ma, T.T.; Shen, Y. Genetic alteration patterns and clinical outcomes of elderly and secondary acute myeloid leukemia. Hematol. Oncol. 2019, 37, 456–463.
- Elessa, D.; Zhao, L.P.; de Oliveira, R.D.; Maslah, N.; Soret-Dulphy, J.; Verger, E.; Marcault, C.; Parquet, N.; Fenaux, P.; Adès, L.; et al. Clinical features and genomic landscape of myeloproliferative neoplasm (MPN) patients with autoimmune and inflammatory diseases (AID). Leukemia 2023, 378, 1741–1744.
- Boddu, P.C.; Zeidan, A.M. Myeloid disorders after autoimmune disease. Best Pract. Res. Clin. Haematol. 2019, 32, 74.
- Rodriguez-Meira, A.; Norfo, R.; Wen, S.; Chédeville, A.L.; Rahman, H.; O’Sullivan, J.; Wang, G.; Louka, E.; Kretzschmar, W.W.; Paterson, A.; et al. Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution. Nat. Genet. 2023, 55, 1531–1541.
- Mendez Luque, L.F.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153.
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-κB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758.
- Karantanos, T.; Teodorescu, P.; Perkins, B.; Christodoulou, I.; Esteb, C.; Varadhan, R.; Helmenstine, E.; Rajkhowa, T.; Paun, B.C.; Bonifant, C.; et al. The role of the atypical chemokine receptor CCRL2 in myelodysplastic syndrome and secondary acute myeloid leukemia. Sci. Adv. 2022, 8, eabl8952.
- Schinke, C.; Giricz, O.; Li, W.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152.
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016, 9, 5023.
- Montes, P.; Bernal, M.; Campo, L.N.; González-Ramírez, A.R.; Jiménez, P.; Garrido, P.; Jurado, M.; Garrido, F.; Ruiz-Cabello, F.; Hernández, F. Tumor genetic alterations and features of the immune microenvironment drive myelodysplastic syndrome escape and progression. Cancer Immunol. Immunother. 2019, 68, 2015–2027.
- Milosevic Feenstra, J.D.; Jäger, R.; Schischlik, F.; Ivanov, D.; Eisenwort, G.; Rumi, E.; Schuster, M.; Gisslinger, B.; Machherndl-Spandl, S.; Bettelheim, P.; et al. PD-L1 overexpression correlates with JAK2-V617F mutational burden and is associated with 9p uniparental disomy in myeloproliferative neoplasms. Am. J. Hematol. 2022, 97, 390–400.
- Palumbo, G.A.; Parrinello, N.L.; Giallongo, C.; D’amico, E.; Zanghì, A.; Puglisi, F.; Conticello, C.; Chiarenza, A.; Tibullo, D.; Di Raimondo, F.; et al. Monocytic Myeloid Derived Suppressor Cells in Hematological Malignancies. Int. J. Mol. Sci. 2019, 20, 5459.
- Vaxevanis, C.K.; Bauer, M.; Subbarayan, K.; Friedrich, M.; Massa, C.; Biehl, K.; Al-Ali, H.K.; Wickenhauser, C.; Seliger, B. Biglycan as a mediator of proinflammatory response and target for MDS and sAML therapy. Oncoimmunology 2023, 12, 2152998.
- Rontauroli, S.; Carretta, C.; Parenti, S.; Bertesi, M.; Manfredini, R. Novel Molecular Insights into Leukemic Evolution of Myeloproliferative Neoplasms: A Single Cell Perspective. Int. J. Mol. Sci. 2022, 23, 15256.
- Han, H.; Byun, J.M.; Shin, D.Y.; Yoon, S.S.; Koh, Y.; Hong, J.; Kim, I.; Lee, C.; Yoo, H.; Yun, H.; et al. Leukemic stem cell phenotype is associated with mutational profile in acute myeloid leukemia. Korean J. Intern. Med. 2021, 36, 401.
- de Castro, F.A.; Mehdipour, P.; Chakravarthy, A.; Ettayebi, I.; Yau, H.L.; Medina, T.S.; Marhon, S.A.; de Almeida, F.C.; Bianco, T.M.; Arruda, A.G.F.; et al. Ratio of stemness to interferon signalling as a biomarker and therapeutic target of myeloproliferative neoplasm progression to acute myeloid leukaemia. Br. J. Haematol. 2023, 204, 206–220.
- Guess, T.; Potts, C.R.; Bhat, P.; Cartailler, J.A.; Brooks, A.; Holt, C.; Yenamandra, A.; Wheeler, F.C.; Savona, M.R.; Cartailler, J.-P.; et al. Distinct Patterns of Clonal Evolution Drive Myelodysplastic Syndrome Progression to Secondary Acute Myeloid Leukemia. Blood Cancer Discov. 2022, 3, 316–329.
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020, 587, 477.
- Taylor, J.; Lee, S.C.; Stanley Lee, C.C.; Sloan Kettering, M. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosom. Cancer 2019, 58, 889–902.
- Kar, S.A.; Jankowska, A.; Makishima, H.; Visconte, V.; Jerez, A.; Sugimoto, Y.; Muramatsu, H.; Traina, F.; Afable, M.; Guinta, K.; et al. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica 2013, 98, 107.
- Stegelmann, F.; Bullinger, L.; Schlenk, R.F.; Paschka, P.; Griesshammer, M.; Blersch, C.; Kuhn, S.; Schauer, S.; Döhner, H.; Döhner, K. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011, 25, 1217–1219.
- Brune, M.M.; Rau, A.; Overkamp, M.; Flaadt, T.; Bonzheim, I.; Schürch, C.M.; Federmann, B.; Dirnhofer, S.; Fend, F.; Tzankov, A. Molecular Progression of Myeloproliferative and Myelodysplastic/Myeloproliferative Neoplasms: A Study on Sequential Bone Marrow Biopsies. Cancers 2021, 13, 5605.
- Benton, C.B.; Boddu, P.C.; DiNardo, C.D.; Bose, P.; Wang, F.; Assi, R.; Pemmaraju, N.; Devendra, K.C.; Pierce, S.; Patel, K.; et al. Janus kinase 2 variants associated with the transformation of myeloproliferative neoplasms into acute myeloid leukemia. Cancer 2019, 125, 1855–1866.
- Stengel, A.; Baer, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Mutational patterns and their correlation to CHIP-related mutations and age in hematological malignancies. Blood Adv. 2021, 5, 4426–4434.
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 Lesions in Leukemic Transformation. N. Engl. J. Med. 2011, 364, 488–490.
- Rampal, R.; Ahn, J.; Abdel-Wahaba, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410.
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Köhne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2160–2168.
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.A.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Gründer, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019.
- Goyal, H.; Chachoua, I.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N. A p53-JAK-STAT connection involved in myeloproliferative neoplasm pathogenesis and progression to secondary acute myeloid leukemia. Blood Rev. 2020, 42, 100712.
- Zarka, J.; Short, N.J.; Kanagal-Shamanna, R.; Issa, G.C. Nucleophosmin 1 Mutations in Acute Myeloid Leukemia. Genes 2020, 11, 649.
- Bahaj, W.; Kewan, T.; Gurnari, C.; Durmaz, A.; Ponvilawan, B.; Pandit, I.; Kubota, Y.; Ogbue, O.D.; Zawit, M.; Madanat, Y.; et al. Novel scheme for defining the clinical implications of TP53 mutations in myeloid neoplasia. J. Hematol. Oncol. 2023, 16, 91.
- Awada, H.; Durmaz, A.; Gurnari, C.; Kishtagari, A.; Meggendorfer, M.; Kerr, C.M.; Kuzmanovic, T.; Durrani, J.; Shreve, J.; Nagata, Y.; et al. Machine learning integrates genomic signatures for subclassification beyond primary and secondary acute myeloid leukemia. Blood 2021, 138, 1885–1895.
- Guglielmelli, P.; Calabresi, L. The MPL mutation. Int. Rev. Cell Mol. Biol. 2021, 365, 163–178.
- Chang, L.; Cui, Z.; Shi, D.; Chu, Y.; Wang, B.; Wan, Y.; Ma, Q.; Zhang, R.; Li, H.; Cheng, X.; et al. Polyclonal evolution of Fanconi anemia to MDS and AML revealed at single cell resolution. Exp. Hematol. Oncol. 2022, 11, 64.
- Lee, C.; Kim, H.N.; Kwon, J.A.; Hwang, J.; Park, J.Y.; Shin, O.S.; Yoon, S.Y.; Yoon, J. Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling. Cancers 2023, 15, 5289.
- Tembrink, M.; Gerding, W.M.; Wieczorek, S.; Mika, T.; Schroers, R.; Nguyen, H.P.; Ben Vangala, D.; Nilius-Eliliwi, V. Novel NUP98::ASH1L Gene Fusion in Acute Myeloid Leukemia Detected by Optical Genome Mapping. Cancers 2023, 15, 2942.
- Zhang, X.; Zhang, Y.; Wang, C.; Wang, X. TET (Ten-eleven translocation) family proteins: Structure, biological functions and applications. Signal Transduct. Target. Ther. 2023, 8, 297.
- Ganster, C.; Müller-Thomas, C.; Haferlach, C.; Strupp, C.; Ogata, K.; Germing, U.; Hildebrandt, B.; Mallo, M.; Lübbert, M.; Müller, C.; et al. Comprehensive analysis of isolated der(1;7)(q10;p10) in a large international homogenous cohort of patients with myelodysplastic syndromes. Genes Chromosom. Cancer 2019, 58, 689–697.
- Hebeda, K.; Boudova, L.; Beham-Schmid, C.; Orazi, A.; Kvasnicka, H.M.; Gianelli, U.; Tzankov, A. Progression, transformation, and unusual manifestations of myelodysplastic syndromes and myelodysplastic-myeloproliferative neoplasms: Lessons learned from the XIV European Bone Marrow Working Group Course 2019. Ann. Hematol. 2021, 100, 117–133.
- Gurban, P.; Mambet, C.; Botezatu, A.; Necula, L.G.; Neagu, A.I.; Matei, L.; Pitica, I.M.; Nedeianu, S.; Chivu-Economescu, M.; Bleotu, C.; et al. Leukemic conversion involving RAS mutations of type 1 CALR-mutated primary myelofibrosis in a patient treated for HCV cirrhosis: A case report. Front. Oncol. 2023, 13, 1266996.
More