Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ryungsa Kim and Version 2 by Fanny Huang.
The common signaling pathways of apoptotic cell death, antiapoptotic pathways, non-apoptotic cell death mechanisms (autophagic, necrotic, and other), signaling pathways involved in the death of drug-sensitive and -resistant tumor cells (with emphasis on c-Jun/activator protein 1 and crosstalk with mitochondrial and endoplasmic reticulum pathways), and therapeutic implications of the modification of signaling pathways leading to cell death (with emphasis on cell death-related gene targeting, interactions of drug resistance factors in drug-resistant cells, and the unfolded protein response pathway).
- signaling pathway
- cell death
- cancer cell
- anticancer drug
1. Introduction
The hallmarks of cancer cells include persistent growth signaling, growth inhibitor evasion, resistance of cell death, unlimited replication capacity, angiogenesis, genomic instability, unleashed phenotypic plasticity, and the evasion of immune destruction [1]. These features allow cancer cells to continue to proliferate, invade neighboring tissues, and spread to other organs to establish metastatic sites. During these processes, the alteration of signaling pathways in cancer cells affects their sensitivity and resistance to anticancer agents and antitumor immunity, making cancer treatment more difficult and less curative.
Anticancer drug-induced cell death can be classified into at least three forms according to morphological and biological criteria: apoptosis, autophagy, and necrosis [2]. Whereas apoptosis and necrosis are irreversible, autophagy is reversible and can lead to cell death or immune escape. The induction of apoptotic cancer cell death is an important component of the therapeutic effects of anticancer drugs [3], and its attenuation correlates with resistance to these drugs [4]. The molecular mechanisms of the intrinsic and extrinsic signaling pathways mediated by mitochondrial outer membrane permeabilization (MOMP) and death receptors (DRs) such as Fas and DR4/5 have been investigated extensively in research on anticancer drug-induced apoptotic cell death [5][6][5,6]. The regulation of these pathways is mediated by proapoptotic and antiapoptotic B-cell lymphoma 2 (Bcl-2) family proteins. Their modulation through the activation of proapoptotic proteins such as Bax and the inhibition of antiapoptotic proteins such as Bcl-2 enhances the therapeutic efficacy against cancer cells [7]. Necrosis and autophagy are also involved in the therapeutic effects of anticancer drugs [8], but the interaction and relationship between apoptotic and autophagic cell death (ACD) in this context remain to be elucidated [9].
Cancer cells’ development of multidrug resistance (MDR) is a consideration in successful cancer treatment. MDR is caused by multiple factors, including the overexpression of transmembrane proteins as drug efflux pumps via ATP-binding cassette transporters such as P-glycoprotein and multidrug resistance-related protein (MRP), increased levels of detoxification enzymes such as glutathione S transferase, the alteration of DNA target enzymes such as topoisomerase I/II, and the attenuation of DNA damage responses and apoptotic signaling pathways [10][11][10,11]. The examination of changes in the signaling pathways leading to apoptosis in drug-resistant cells provides a deeper understanding of the molecular mechanisms underlying the therapeutic effects of anticancer drugs.
Transcription factors such as activator protein 1 (AP-1) and p53 play important roles in the signaling pathway leading to apoptotic cell death [12]. Activated c-Jun N-terminal kinase (JNK) phosphorylates c-Jun, which heterodimerizes with the c-Fos family as AP-1 and activates DNA damage-inducible genes [growth arrest- and DNA damage-inducible gene 153 (Gadd153)/CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP)], Bak, Bim, and p53 to promote apoptotic cell death [13]. The attenuation or alteration of signaling pathways involved in transcription factor-mediated apoptotic cell death can lead to drug resistance in cancer cells [14]. The attenuation of the DNA damage response by anticancer drugs leads to a decrease in antitumor immune activity due to a reduction in immunogenic cell death (ICD). The induction of ICD activates the ICD signaling pathway by releasing damage-associated molecular patterns (DAMPs) from dying tumor cells, leading to the activation of tumor-specific immune responses, and providing for the long-term efficacy of anticancer drugs [15][16][15,16].
2. Common Pathways of Apoptotic Cell Death
2.1. Intrinsic (Mitochondrial) Pathway
The signaling pathways induced by anticancer drugs are summarized in Figure 1. Mitochondria play a pivotal role in the regulation of anticancer drug-induced apoptotic cancer cell death. In the intrinsic pathway, increased MOMP leads to the release of molecules such as Cyt c, second mitochondria-derived activator of caspase (Smac)/direct inhibitor of apoptosis binding protein with low isoelectric point (DIABLO), and Omi/HtrA2 from the inner mitochondrial space and the activation of a caspase cascade via the activation of the proapoptotic protein Bax/Bak [17][18][19][20][21][17,18,19,20,21]. Cyt c activates caspase 9/3 via apoptosomes composed of apoptotic peptidase activating factor 1 (Apaf-1) and procaspase 9 in the presence of deoxy-ATP or ATP [22]. Smac/DIABLO and Omi/HtrA2 activate the caspase cascade by inhibiting the inhibitor of apoptosis protein (IAP), leading to apoptotic cell death [18][19][20][18,19,20]. p53 regulates the transcription of downstream proapoptotic target genes such as Bax, Noxa, Puma, and Fas and binds to antiapoptotic proteins such as Bcl-2 and Bcl-xL to increase Bcl-2 homology domain 3 (BH3)-only proteins such as Bid and Bim, thereby regulating the Bax/Bad-mediated apoptotic cell death pathway [23]. Bcl-xS inhibits Bcl-xL, resulting in the activation of the Bax/Bak-mediated pathway [24].
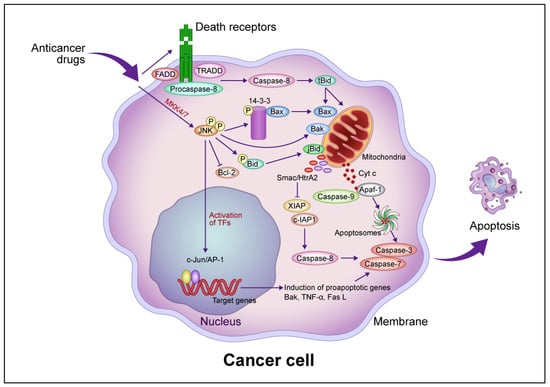
Figure 1. Common intrinsic and extrinsic apoptotic signaling pathways induced by anticancer drugs. In the intrinsic pathway, anticancer drugs activate JNK via mitogen-activated protein kinase kinase (MKK)4/7, which phosphorylates the 14-3-3 protein, dissociates Bax, and activates Bak. Bax and Bak translocate to the mitochondrial outer membrane and release Cyt c. Cyt c then forms apoptosomes containing apoptotic protease activating factor 1 (Apaf-1) and procaspase-9, activating the caspase cascade that leads to apoptotic cell death. JNK phosphorylates Bid (jBid) and migrates through the mitochondria to release second mitochondria-derived activator of caspases (Smac)/high temperature requirement A2 (HtrA2), which inhibits the X-linked inhibitor of apoptosis protein (XIAP) and cellular inhibitor of apoptosis protein (c-IAP), resulting in the activation of caspase-8 and the caspase cascade. JNK also phosphorylates B-cell lymphoma 2 (Bcl-2) and inhibits its function. In the extrinsic pathway, anticancer drugs activate death receptors that recruit the Fas-associated death domain (FADD), tumor necrosis factor (TNF) receptor-associated death domain (TRADD), and procaspase-8 to form a death-induced signaling complex, which in turn activates caspase-8. Caspase-8 cleaves, generating truncated Bid (tBid), which translocates to the mitochondria, releasing proapoptotic proteins such as Cyt c and Smac/HrtA2. JNK activates c-Jun/activator protein 1 (AP-1), which induces apoptosis-promoting genes such as Bak, TNF-α, and Fas L, in turn activating the caspase cascade leading to apoptotic cell death. Abbreviation: TF, transcription factor. This figure was custom-made by Wiley Editing Services based on researchers' freehand drawing.
JNK is required for the release of Cyt c from mitochondria in apoptotic cell death [25]. Activated JNK promotes the dissociation of Bax from this protein by translocation from the cytosol to the mitochondria via phosphorylation of 14-3-3, the cytoplasmic anchor of Bax [26]. Mouse embryonic fibroblasts (MEFs) derived from JNK1−/− JNK2−/− mice resist apoptosis in response to diverse genotoxic and cytotoxic stresses, providing evidence that JNK is involved in apoptotic signaling [27][28][27,28]. Growth factor-induced antiapoptotic JNK activation is rapid and transient, whereas γ-ray-induced proapoptotic JNK activation is delayed [29]. JNK activation by anticancer drugs is sustained long term in drug-sensitive cells and transient in drug-resistant cells [30]. The transfection of a dominant-negative JNK allele inhibited JNK activity and blocked anticancer drug-induced apoptosis in drug-sensitive cells [30].
JNK contributes to the phosphorylation of p53 family proteins in the apoptosis signaling pathway [31], which likely involves the p53-mediated upregulation of proapoptotic genes such as Bax and Puma [32]. JNK activation induced by DNA damage also stabilizes and activates p73, a member of the p53 family that induces genes such as Bax and Puma [33][34][33,34]. Cisplatin-induced p73-mediated apoptosis requires JNK, which phosphorylates p73. Mutations at the p53 phosphorylation site of JNK inhibit p73 stabilization by cisplatin and reduce p73 transcriptional activity, thereby reducing cisplatin-induced apoptosis [33]. JNK induces the expression of proapoptotic genes and decreases the expression of prosurvival genes through multiple transcription factors in a cell type- and stimulus-specific manner.
2.2. Extrinsic (Death Receptor-Mediated) Pathway
MOMP induced by Bax/Bak activation promotes apoptotic cell death, and Bid activates Bax/Bak following activation by caspase-8. Caspase-8, in turn, is activated by the binding of Fas and DR4/5 to the death factor receptor Fas L and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which recruits the tumor necrosis factor receptor 1 (TNFR1) death domain protein (TRADD), Fas-related death domain protein (FADD), and procaspase-8 and forms an intracellular death-induced signaling complex (DISC) that activates procaspase-8 [35]. Following TRAIL activation, FADD is recruited after TRADD dissociates and forms complexes with the receptor-interacting protein (RIP) and tumor necrosis factor receptor–associated factor 2 (TRAF2), which mediate cell survival and death through nuclear factor–kappa B (NF-κB) and JNK1, respectively. Caspase-8 proteolytically cleaves Bid to form tBid, activating the Bax/Bak-mediated mitochondrial pathway [36]. TNF-α activates caspase-8, which induces JNK to activate Bid (jBid) through phosphorylation-mediated cleavage and promote the release of Smac and Omi [37]. The inhibition of cellular IAP 1 by Smac and X-linked inhibitor of apoptosis protein (XIAP) by Smac and Omi leads to the activation of the execution factors caspases-3 and -7, leading to apoptosis [38]. Caspase-8 directly activates caspase-3 without amplification of the mitochondrial pathway [39] but also induces lysosome-associated non-apoptotic cancer cell death [40].
JNK is activated by cisplatin treatment, and its sustained activation induces c-Jun activation, in turn stimulating Fas L, a downstream gene associated with apoptosis, in sensitive ovarian cancer cells [41]. The inhibition of cisplatin-induced JNK activation prevents this form of apoptosis, and this activation is transient in drug-resistant cells [41]. Stimulation by the selective adenovirus-mediated delivery of mitogen-activated protein kinase kinase 7 (MKK7) or MKK3, upstream activators of JNK, reactivated Fas L expression and increased the susceptibility of resistant cells to apoptotic cell death [41]. TNF-α induced apoptotic cell death in breast cancer cells and mouse fibroblasts via JNK activation, despite the absence of an antiapoptotic inhibitor of the nuclear factor–kappa B (I-κB)/NF-κB pathway, and the inhibition of JNK activation suppressed this process [42]. The duration of JNK activation by anticancer drugs may be important for the induction of apoptotic cell death.
2.3. Antiapoptotic Pathway
The release of apoptotic small molecules by MOMP via Bax/Bak is an essential event in the caspase-dependent and caspase-independent apoptotic pathways and is inhibited by antiapoptotic proteins such as Bcl-2 and Bcl-xL [43]. These proteins inhibit the migration and oligomerization of Bax before it is inserted into the mitochondrial outer membrane. Bcl-xL inhibits DISC formation and Bid activation by caspase-8, suggesting that it regulates not only the mitochondrial pathway but also the upstream receptor-dependent pathway [44]. Bcl-2 partially inhibits DR-dependent pathways. MOMP proceeds via the loss of mitochondrial membrane potential, which depends on the death trigger and generates reactive oxygen species (ROS) that in turn activate lysosomal enzymes involved in non-apoptotic cell death [45]. Bcl-2 and Bcl-xL prevent the loss of mitochondrial membrane potential and subsequent production of ROS, partially through the antioxidant function of Bcl-2 [44].
The antiapoptotic phosphoinositide 3-kinase (PI3K)/Akt pathway plays important roles in tumor development and progression [46]. Akt is a serine-threonine kinase composed of three homologous proteins and is activated by hormones and growth factors. It regulates apoptosis-promoting proteins such as Bax and Bad; Bax is phosphorylated to promote heterodimerization with myeloid leukemia cell 1 (MCL1) and Bcl-xL, which inhibits its translocation to mitochondria, and Bad is dephosphorylated to bind and inactivate 14-3-3 [47][48][47,48]. Akt also regulates Bcl-2 expression via cAMP response element binding protein (CREB) and directly inhibits caspase-9 [49][50][49,50]. It inhibits p53 function via the activation of murine double minute 2 (MDM2) [51]. I-κB is phosphorylated by Akt and activates NF-κB as an inhibitor of apoptosis [52]. NF-κB activates important antiapoptotic proteins such as Bcl-xL, XIAP, and cellular FLICE-inhibitory protein [53]. It inhibits p27 and induces ABCB1 (MDR1) and matrix metalloproteinase-9, which is involved in cancer cell cycle regulation, drug resistance, and metastasis [54][55][56][54,55,56]. NF-κB prevents TNF-α-induced apoptotic cell death by inhibiting the JNK cascade, including the caspase inhibitor XIAP [57], via its antioxidant function, which reduces TNF-α-induced ROS accumulation. The antiapoptotic activity of NF-κB is regulated by the inhibition of ROS accumulation and the regulation of JNK cascade activation [53].