Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria Camila Trillos-Almanza and Version 3 by Rita Xu.
Branched-chain amino acids (BCAAs), indispensable for protein synthesis and metabolic pathways, undergo unique tissue-specific processing in skeletal muscle and liver. The liver, responsible for amino acid metabolism, plays a distinctive role in sensing BCAAs catabolism, influencing glucose regulation and contributing to the systemic metabolism of BCAAs.
BCAAs, indispensable for protein synthesis and metabolic pathways, undergo unique tissue-specific processing in skeletal muscle and liver. The liver, responsible for amino acid metabolism, plays a distinctive role in sensing BCAAs catabolism, influencing glucose regulation and contributing to the systemic metabolism of BCAAs.
- amino acids
- branched-chain
- chronic liver disease
- muscle–liver crosstalk
1. Introduction
The branched-chain amino acids (BCAAs) Isoleucine (Ile), Leucine (Leu) and Valine (Val), represent around 35% of the total twelve essential amino acids in most mammals with a relative abundance of approximately 1.6:2.2:1.0 (Val:Leu:Ile), with Leu being the most abundant and Ile the least abundant [1]. BCAAs are branched, small and hydrophobic molecules, that are used for protein synthesis [2] and derived from nutrition, since they cannot by synthesized by the body itself. In general, BCAAs share metabolic routes and they are usually present in the same dietary sources and therefore metabolized together, which is also the reason why they are usually studied in combination [3].
BCAAs are not only essential substrates for protein synthesis but also regulate energy metabolism, for example, by contributing to gluconeogenesis and lipid metabolism [4]. For instance, in muscle, BCAAs provide non-specific carbons as a source for oxidation during energy production, yielding energy more efficiently than glucose [5]. In addition, BCAAs stimulate protein synthesis by enhancing mRNA translation and by favoring protein turnover [6].
The liver is responsible for BCAAs oxidation using metabolic BCAAs coming from skeletal muscle (and other tissues), to maintain protein turnover, amino acid levels and energy homeostasis (e.g., gluconeogenesis), among others [7]. Whereas most amino acids are metabolized in the liver, skeletal muscle plays a pivotal role in BCAAs metabolism, actively participating in their utilization and significantly impacting diverse physiological processes. Skeletal muscle has a high activity of the enzyme branched-chain aminotransferase 2 (BCAT2), the first enzyme involved in BCAAs catabolism which is barely expressed in the liver [8].
Skeletal muscle protein synthesis and glucose uptake are stimulated by BCAAs through the promotion of the translocation of glucose transporters to the plasma membrane [9], while proteolysis is suppressed by BCAAs [10]. Additionally, BCAAs activate metabolic signaling pathways like the mTOR and PI3K-Akt pathways, ultimately leading to insulin resistance [4][11][4,11]. Recent studies suggest that BCAAs have a role in glucose homeostasis and it has been reported that BCAAs plasma levels correlate with obesity, glucose intolerance, insulin resistance and increased risk of developing type 2 diabetes [12]. In addition, it has been reported that the liver, as a regulator of BCAAs catabolism, plays an important role in the interplay between BCAAs metabolism and energy homeostasis [13]. Moreover, defective BCAAs catabolism results in impaired glucose metabolism [14].
It has also been suggested that BCAAs might be involved in the pathophysiology of chronic liver diseases (CLD). Impairment of BCAAs catabolism has been associated with liver fibrosis [15][16][15,16]. A negative correlation between fibrosis stage and plasma concentrations of BCAAs has been described, primarily attributed to the decrease in serum BCAAs levels to compensate for the impaired hepatic urea cycle [15]. BCAAs catabolism alterations might be mainly influenced by the suppression of the enzymatic activity of the first two enzymes in their catabolic pathway, BCAAs aminotransferase (BCAT) and branched-chain α-keto acid dehydrogenase (BCKD). Therefore, these enzymes may be suitable therapeutic targets for the prevention of CLD [17][18][17,18]. The molar ratio of BCAAs residues (Leu, Ile and Val) to aromatic amino acid (AAA) residues (Tyr, Trp and Phe) is known as the Fischer ratio. A low Fischer ratio, indicative of disrupted amino acid metabolism, is associated with the development of hepatic encephalopathy (HE), sarcopenia and hepatocarcinogenesis [18] and increased mortality in advanced CLD [19]. The Fischer ratio serves as a valuable predictor of adverse clinical events, emphasizing its importance in assessing the severity of liver dysfunction and guiding therapeutic strategies. Furthermore, BCAAs supplementation is also effective against fibrosis by downregulating hepatic stellate cell activation and TGF-B signaling as a consequence of mTORC1 activation [15].
2. Metabolism of BCAAs: Interplay between Skeletal Muscle and Liver
2.1. BCAAs Transamination
Mammals cannot synthesize BCAAs, but bacteria, fungi and plants can and use threonine (for Ile) and pyruvate (for Val and Leu) as precursors [1]. In general, BCAAs account for approximately 15% of the total amount of amino acids in human muscle protein and they are the main nitrogen source for glutamine and alanine synthesis in skeletal muscle tissue [2]. The first step in BCAAs catabolism, which occurs in all organisms, is the (reversible) transamination to branched-chain α-ketoacids (BCKAs) by mitochondrial BCATs (Figure 1) [17].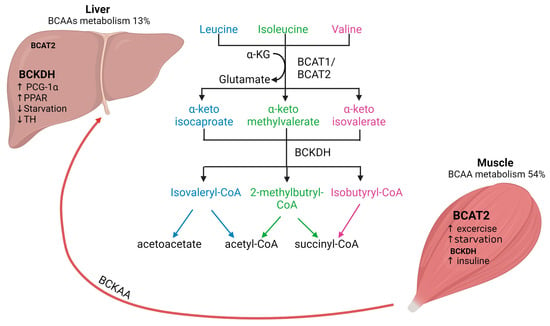
Figure 1. Interplay of BCAAs metabolism between human skeletal muscle and liver. Arrows represent the metabolic steps involved in the conversion and utilization of BCAAs. BCAAs, branched-chain amino acids; BCAT, branched-chain aminotransferase; BCKDH, branched-chain alpha-keto acid dehydrogenase.
2.2. BCAAs Oxidation
The second step of BCAAs catabolism is oxidative decarboxylation (irreversible) catalyzed by the BCKDH to produce branched-chain acyl-CoA esters (Figure 1) [17]. The activity of the BCKDH enzyme is high in the liver, brain, and kidney but low in skeletal muscle [7]. Oxidative decarboxylation by BCKDH is performed mainly in the liver using BCKAs produced in the skeletal muscle, which are imported into the liver [23], implying an interplay between skeletal muscle and liver in BCAAs metabolism. However, overall skeletal muscle accounts for 54% of total BCAAs catabolism compared to only 13% in the liver [2]. The final step involves the catabolism of the branched-chain acyl-CoA esters, which differs for each BCAAs (Figure 1). Leu is converted into acetyl-CoA and acetoacetate production (ketogenic), Val into succinyl-CoA (glucogenic) and Ile into acetyl-CoA and succinyl-CoA. All these metabolites feed into the citric acid cycle (TCA) contributing to energy generation [1]. Furthermore, BCAAs conversion to BCKAs in skeletal muscle produces glutamate, which is combined with pyruvate for the production of alanine and released for its transport to the liver. Alanine is further used as a substrate for glucose generation through gluconeogenesis and, finally, this glucose is taken up by the skeletal muscle for energetic metabolism [24][25][24,25]. The interaction between skeletal muscle and liver by BCAAs trafficking is necessary for energy homeostasis. The regulation of BCAAs is not completely understood yet. BCAT activity is physiologically upregulated with exercise and food intake but downregulated by starvation. In skeletal muscle, BCTA2 activity can be upregulated by exercise, which has been demonstrated by the increase of KIC (2-keto-isocaproate/4-methyl-2-oxopentanoic acid), a subproduct of leucine transamination [2]. Likewise, endurance training has been shown to increase BCAT2 expression in muscle and therefore increases BCAAs oxidation [22]. Physiological regulation of BCTA2 expression seems to be controlled by transcription factors such as PGC-1α factor, which leads to an increase in BCAT expression, likely due to higher mitochondrial activity and via glucocorticoid receptor (GR) [2]. Regarding food intake, it has been shown that BCAAs oxidation is increased after feeding and decreased upon caloric restriction. Interestingly, if fasting is prolonged (starvation), BCAAs oxidation increases again as a feedback mechanism to provide substrates for gluconeogenesis in the liver [26]. In addition, BCAAs oxidation is affected by several factors, e.g., insulin increases BCAAs oxidation [24]. It has been reported that BCKDH is regulated by allosteric inhibition by subproducts (e.g., NADH, α-ketoisocaproate, branched acyl-CoA esters), but it is also regulated by phosphorylation [27]. Regarding BCAAs catabolism in the liver, it is known that BCKDH catabolic enzymes are also positively regulated by transcriptional factors such as PCG-1α and PPAR transcription factor family members in the liver. In contrast, BCKDH activity is decreased by thyroid hormone and during starvation [24]. Furthermore, it is known that BCKAs oxidation might also affect liver metabolism and glucose homeostasis [14]. For instance, BCKAs accumulation caused by decreased BCKA oxidation results in gluconeogenesis inhibition. This interplay between BCAAs and liver glucose metabolism might be explained by the role of the liver as a sensor of BCKAs circulating levels and of BCAA catabolism [13]. Moreover, recent evidence suggests that BCKDH activity can decrease during metabolic alterations (e.g., obesity) leading to a shift in BCKA oxidation from the liver to skeletal muscle and resulting in insulin resistance [28]. These mechanisms might explain in part the evidence of the association between BCAAs and metabolic dysfunction. In fact, increased levels lead to metabolic alterations through mechanisms such as mitochondrial dysfunction and insulin resistance by the activation of the mTOR signaling pathway [29]. Moreover, it has been shown that BCAAs catabolism alterations are associated with the development of CLD [16]. In summary, the catabolism of BCAAs involves intricate processes primarily in the liver, yet significantly influenced by skeletal muscle activity. Regulatory mechanisms, such as exercise, starvation and transcription factors, are fundamental in this process. Dysregulation of BCAAs catabolism may precipitate metabolic dysfunction, underscoring its relevance to CLD.3. Clinical Insights into CLD: Role of BCAAs in Musculoskeletal Health
3.1. BCAAs Concentration in Patients with CLD
In advanced stages of cirrhosis, several factors contribute to a poor nutritional state, including low nutrient intake, altered metabolism, impaired absorption, impaired digestion, and rapid satiety. A deficient nutritional status is prevalent among adults with CLD, with reports indicating rates from 5% to 74%, depending on the assessment method used [30]. Among these patients, up to 59% exhibit moderate to severe malnutrition, with alcohol-associated liver disease (ALD) being the most prevalent etiology [31]. As the disease progresses, extended fasting periods and insulin resistance prompt muscle cells to initiate gluconeogenesis. This process utilizes BCAAs as a source for synthesizing glutamine and alanine. Furthermore, skeletal muscles play a role in detoxifying ammonia by transforming ammonia and glutamate into glutamine by glutamine synthetase, a reaction that utilizes BCAAs as the primary nitrogen source. These mechanisms, coupled with malnutrition, collectively contribute to the observed plasmatic depletion of BCAAs in patients with CLD. It has been described that healthy subjects typically exhibit total BCAAs plasma concentrations ranging from 423 μmol/kg to 646 μmol/kg, while patients with cirrhosis of different etiologies (ALD, viral hepatitis, metabolic dysfunction-associated steatotic liver disease (MASLD)) and HCC often demonstrate lower levels, ranging from 278 μmol/kg to 535 μmol/kg across various studies. The studies by Montanari A. et al. [32] and Dam G. et al. [33][34][33,34] reported statistically significant differences in Leu and Val concentrations between adults with CLD and healthy individuals. The intracellular levels of BCAAs in muscle and their uptake into muscle are also compromised. Dam G. et al. [33][34][33,34] demonstrated a consistent decrease in BCAAs uptake in adults with CLD (67 μmol/min) compared to controls (110 μmol/min). Montanari A. et al. [32] reported a substantial depletion of Val levels in muscle tissue among adults with CLD. Although Val was significantly reduced (222 umol/kg vs. control (368 umol/kg), a comparable declining trend was observed with Leu and Ile levels. The decline in nutritional status correlates with the progression of liver diseases. Janota B. et al. [35][40] investigated 118 adults with CLD and demonstrated a strong correlation between the Child-Pugh (CP) score and nutritional status. This deterioration primarily affected muscle mass, falling below standard values in 1.9% (CP score A), 29.4% (CP score B), and 56.2% (CP score C) of cases. Trillos-Almanza. et al. [36][38] reported a correlation between plasma BCAAs concentrations and the CP score, showing reduced BCAAs levels in more severe cases (CP score B and C) compared to the less severe category (CP score A). In conclusion, the intricate relationship between CLD, nutritional status, and BCAAs highlights the need for comprehensive nutritional interventions in the management of these patients. Understanding these complex interactions could pave the way for targeted nutritional strategies aimed at improving the wellbeing of individuals with end-stage liver disease, particularly in addressing the challenges associated with malnutrition and BCAAs depletion.3.2. Correlation of Muscle Health and BCAAs in Patients with CLD
The relation between nutritional status, muscle functionality (measured by handgrip strength), and muscle mass (determined by the skeletal muscle index (SMI)) is connected to the development of sarcopenia as demonstrated by Sehgal P. et al. [37][41]. Sarcopenia is currently recognized as muscle failure, defined by a combination of factors such as diminished skeletal muscle quantity, increased fat accumulation within the muscle, reduced muscle strength, compromised physical performance, and alterations in circulating biological markers [38][42], with low strength as the key characteristic for the condition [39][43]. In a multicenter study evaluating patients with cirrhosis of diverse etiologies, the prevalence of sarcopenia was 45.4% to 70% [40][44]. Notably, this prevalence is considerably higher when compared to the 10–16% range observed in the elderly population [41][45]. Regardless of the nutritional status, patients with CLD experience reduced skeletal muscle strength and mass. Skeletal muscle mass requires the analysis of a group of muscles within a single anatomic area using one or more techniques such as dual-energy X-ray absorptiometry (DXA), bioelectrical impedance analysis (BIA), CT scan or creatinine excretion. Muscle strength evaluation employs dynamometers for isometric strength and measures power and torque for isokinetic strength. Complementary physical performance tests, including gait speed, chair stands, balance tests, the 400 m walk test, and the 6 min walk test, are commonly employed [42][46]. The association between lower BCAAs levels and diminished muscle strength and functionality has been highlighted by Trillos-Almanza et al. [36][38]. In a cross-sectional study, 92 CLD patients were submitted to different functional tests that evaluate mobility, agility and muscle strength of different group of muscles. Hand grip strength demonstrated associations with Leu and Ile in males. In more complex tests that evaluate strength, coordination and balance, such as the Timed Up and Go (TUG) and sit-to-stand test, there was an inverse relationship between the test performance and total BCAAs. These findings underscore the association of BCAAs with liver disease severity and impaired muscle function. Xiang Q. et al. [43][35] corroborated these findings by analyzing hand grip strength in 127 CLD patients with low BCAAs levels, observing a decrease in performance (handgrip strength ≤ 25.47 ± 5.84 kg) and diagnosing sarcopenia in 37 of these patients. These findings underscore the association of BCAAs with liver disease severity and impaired muscle function.3.3. Interplay of BCAAs, mTOR Signaling, Ammonia, and Mitochondrial Dysfunction in the Skeletal Muscle during CLD
The mechanisms underlying muscle mass loss and functionality have been extensively studied. Skeletal muscle mass equilibrium depends on factors such as protein intake, synthesis, and degradation. Many factors contribute to low nutrient intake, including dietary restrictions, altered satiety (linked to low ghrelin and high leptin plasma levels [44][47], substance abuse (opioids, alcohol), and HE. Regarding protein degradation, this one is impaired in chronic liver disease since endoplasmic stress activates the ubiquitin-proteasome system (UPS), which cause protein misfold and autophagy, a cellular process crucial for the degradation and recycling of cellular components. On the other hand, among the key signaling pathways associated with nutritional and metabolic status is the mTOR pathway. BCAAs play a crucial role in protein synthesis and mTOR1 signaling. mTOR signaling is crucial due to its role in regulating various metabolic processes, including ureagenesis. Impaired mTOR signaling disrupts ureagenesis, leading to increased ammonia levels, particularly notable in adults with cirrhosis and sarcopenia where venous ammonia levels are elevated [45][48]. Elevated ammonia levels can prompt increased ammonia uptake in muscles, resulting in the overactivation of the TCA cycle. This overactivation, in turn, reduces mitochondrial ATP synthesis, ultimately triggering a senescence-associated secretory phenotype in skeletal muscle. This phenomenon is mediated by sirtuin-mediated deacetylation [46][47][49,50]. Furthermore, it contributes to heightened oxidative stress within the muscles, as suggested by Davuluri, G. et al. [48][51]. Hence, maintaining functional mTOR signaling helps to regulate these processes and BCAAs may play a role in mitigating the adverse effects associated with increased ammonia levels and oxidative stress in skeletal muscles. Skeletal muscle from patients with low BCAAs levels and cirrhosis reveals reduced the activation of downstream targets of mTOR1 (p70S6K, S6, 4EBP1) but elevated levels of the intracellular amino acid sensor General Control of Nutrition Derepressed 2 (GCN2) [49][37]. This suggests activation of autophagic pathways, as GCN2 is known to elevate autophagic activity in response to short-term essential amino acid deprivation, particularly leucine, in an mTOR1 dependent manner [50][52]. Additionally, ammonia levels among patients with CLD range from 57.7 ± 30.1 µg/dL [51][36] to 125 ± 50 µg/dL [32], while healthy adults maintain concentrations between 19–94 µg/dL, varying between genders [52][53]. This high ammonia concentration correlates with what was found by Dam G. et al. [33], who demonstrated lower BCAAs uptake in leg muscles in adults with CLD (healthy: 196 ± 67 µmol/L vs. cirrhosis: −84.7 ± 110 µmol/L), with those of ALD etiology exhibiting the most pronounced effects [34]. This diminished uptake adversely affects muscle ammonia detoxification. Ammonia serves as a stimulant for proteolysis in adults with CLD by initiating NF-κB-mediated transcription of myostatin [53][54]. Elevated myostatin levels, triggered by ammonia, further escalate proteolysis via Akt inhibition and autophagy activation [54][55]. Conversely, BCAAs are known to inhibit proteolysis [55][56][56,57], and one of the mechanisms involved is ubiquitin-proteasome signaling involving TRIM63 (MURF1) and FBXO32 (Atrogin-1) [57][58]. In a study by Tsien C. et al. [49][37] involving muscle samples from ALD cirrhotic patients (n = 6), increased myostatin protein were observed, accompanied by BCAAs depletion. Finally, high ammonia increases the TCA cycle causing mitochondrial dysfunction in muscle cells. In this matter, Doi J. et al. [58][39] studied patients (n = 8) with cirrhosis of viral etiology and a observe low (0.68–2.31) Fischer´s ratio (BCAAs/AAAs) (normal ≥ 3) together with a significantly change in the intracellular pH (from 7.35 to 0.49 ± 0.16 compared to 0.20 ± 0.18 in healthy individuals) and the creatine phosphate index (0.55 ± 0.12 compared to 0.35 ± 0.19 in the healthy volunteers) in skeletal muscle samples. Both of these changes indicate an overwork in energy production and metabolism, suggesting an indirect link between low BCAAs and altered mitochondrial function, which could explain the impaired protein synthesis and muscle functionality. Taken together, these findings suggest that BCAAs depletion, along with alterations in ammonia metabolism, myostatin activation, and disrupted mTOR signaling, significantly impact muscle mass, functionality, and overall nutritional status in patients with liver diseases (Figure 2). These multifaceted relationships underscore the complex interplay between liver function, muscle metabolism, and nutritional status in these individuals. While further research and validation studies are necessary to establish the value of BCAAs levels as prognostic markers, the strong correlations observed in multiple studies indeed suggest their potential utility in staging nutritional status and identifying sarcopenia in patients with CLD.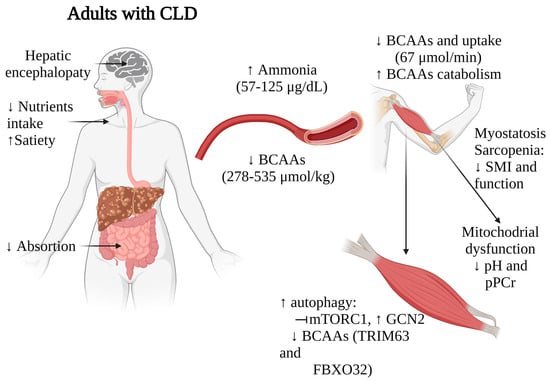
Figure 2. Impact of BCAAs alterations on muscle functionality in adults with CLD. Arrows indicate increase (↑) or decrease (↓). CLD, chronic liver disease; BCAAs, branched-chain amino acids.