胃癌被列为全球第五大最普遍的癌症,长期以来一直是许多人热烈讨论的话题。然而,近年来,社会上胃癌的发病率并没有下降,反而呈逐渐上升的趋势。十多年来,胃癌的治疗效果一直没有明显改善。这归因于癌症的异质性,这使得流行的靶向治疗无效。蛋氨酸是一种必需氨基酸,许多研究表明它与胃癌的发展有关。Gastric cancer is ranked as the fifth most prevalent cancer globally and has long been a topic of passionate discussion among numerous individuals. However, the incidence of gastric cancer in society has not decreased, but instead has shown a gradual increase in recent years. For more than a decade, the treatment effect of gastric cancer has not been significantly improved. This is attributed to the heterogeneity of cancer, which makes popular targeted therapies ineffective. Methionine is an essential amino acid, and many studies have shown that it is involved in the development of gastric cancer. Our study aimed to review the literature on methionine and gastric cancer, describing its mechanism of action to show that tumor heterogeneity in gastric cancer does not hinder the effectiveness of methionine-restricted therapies. This research also aimed to provide insight into the inhibition of gastric cancer through metabolic reprogramming with methionine-restricted therapies, thereby demonstrating their potential as adjuvant treatments for gastric cancer.
- gastric cancer
- methionine
- helicobacter pylori
- programmed cell death
- immune response
- gastric cancer
1. 引言
1.Introduction
In China, gastric cancer ranks second in terms of incidence among all types of cancer, and it also holds the second-highest cancer-related mortality rate. Compared to most developed countries, China exhibits a higher mortality-to-incidence ratio (0.845) and a 5-year prevalence rate of 27.6 cases per 100,000 people [1]. Of even more significant concern is the escalating prevalence of early-onset gastric cancer (EOGC) in the young Chinese population [2].
There are many risk factors for gastric cancer [3], including Helicobacter pylori infection [4], smoking [5–7], dietary habits, abdominal obesity [8], alcoholism [9], and genes and genetics [9]. In China, with the improvement of sanitary conditions and the importance people attach to health, the incidence of gastric cancer was reduced by a decrease [10,11] in the prevalence of Helicobacter pylori infection [12], a decrease in preserved foods [13], etc. However, due to changes in living and working environments, as well as shifts in lifestyle, new factors contributing to the development of cancer have emerged [14]. Factors such as heightened work pressure, abdominal obesity, and unhealthy dietary habits (such as reliance on takeaways, processed meats, barbecue, fried food, and late-night snacks) have been identified as potential contributors to the increased incidence of gastric cancer [1,15–18]. Remarkably, these factors have shown a significant impact on the younger population (below 50 years old) in China. This particular form of gastric cancer is commonly referred to as early-onset gastric cancer [19]. It is believed to be linked to gastritis, the dysbiosis of the gastrointestinal microbiota, and the heightened utilization of antibiotics and acid suppressants [20]. According to anatomical location, gastric cancer can be categorized broadly as cardia gastric cancer, non-cardia gastric cancer, and gastroesophageal junction cancer. Likewise, based on the Lauren classification, gastric cancer can be classified into two main types: diffuse type and intestinal type [21]. According to the 2014 Cancer Genome Atlas Project, gastric cancer can be categorized into four subtypes: Epstein–Barr virus (EBV) type, microsatellite instability (MSI) type, chromosomal instability (CIN) type, and genome-stable type. These subtypes exhibit varying etiological and pathogenic mechanisms, which, in turn, have implications for treatment strategies and prognostic outcomes [22].
However, despite the increasing research being conducted on gastric cancer, the treatment outcomes of gastric cancer have not significantly improved from their previous states over a decade ago [23]. Early non-metastatic gastric cancer is primarily treated with endoscopic treatment or surgical resection [24]. For non-EGJ gastric cancer patients, the current standard treatment is D2 gastrectomy followed by adjuvant chemotherapy. Neoadjuvant therapy can be considered for patients with resectable advanced gastric cancer (C ⅲ or above) [25]. For patients with advanced EGJ gastric cancer, neoadjuvant chemoradiotherapy is an option. Targeted therapies approved for the treatment of gastric cancer include trastuzumab as the first-line treatment for HER2-positive patients, ramucirumab as the second-line treatment for anti-angiogenesis, and nivolumab or pembrolizumab as the third-line treatment for anti-PD-1 [26]. Perioperative or adjuvant chemotherapy can improve the survival rate of patients with stage 1b cancer or higher [27]. Sequential chemotherapy, starting with platinum and fluoropyrimidine [28], is used for advanced gastric cancer. However, the median survival time remains less than 1 year. Additionally, many phase II and phase III clinical trials of molecular targeted drugs have failed. After analyzing the reasons, it was concluded that the heterogeneity of molecular characteristics among cancer cells in patients makes the efficacy of molecular targeted drugs highly unstable [29–36]. This suggests that the use of molecular targeted drugs in the treatment of gastric cancer may not be an ideal pathway. It is therefore an urgent problem to find a new approach to treat gastric cancer that can mitigate the influence of heterogeneity.
Methionine (Met) is one of the essential amino acids in the human body that cannot be synthesized in vivo. Numerous studies have demonstrated [37] the dependence of gastric cancer cells on Met, and an environment lacking Met is conducive to the efficacy of chemotherapy for gastric cancer. Furthermore, the γMetase enzyme can inhibit the proliferation of gastric cancer cells by cleaving methionine [38]. This suggests that Met may stimulate gastric cancer cell proliferation. Additionally, Met can undergo direct conversion to S-adenosyl methionine (SAM) through adenylyltransferase, which serves as a crucial direct methyl donor in the body. SAM plays a significant role in methylation processes, contributing methyl groups to over 50 substances within the body [39]. Therefore, Met can influence the progression of gastric cancer through various pathways. On the one hand, the conversion of Met to SAM can reverse proto-oncogene hypomethylation and subsequently decrease their expression, thereby inhibiting the proliferation of gastric cancer cells [40–43]. It has also been suggested that SAM is associated with ferric death in gastric cancer [44].
On the other hand, the overexpression of Metase significantly promotes apoptosis and autophagy, thereby inhibiting gastric cancer [40,45,46]. Additionally, Met can exert an impact on the development of gastric cancer by influencing the survival of Helicobacter pylori [47–50]. It has been shown that Met enhances the efficacy of the chemotherapeutic drugs 5-fluorouracil and Cisplatin [47,51–53]. The objective of this review is to analyze the role and mechanism of Met in the development of gastric cancer and to consider its potential implications in the treatment of gastric cancer.
- The Relationship between Met and the Methionine Cycle
Met is an essential amino acid, and its metabolism is governed by the core cycle known as the methionine cycle. In this cycle, Met undergoes a conversion to SAM through the action of Met adenosyltransferase (MAT). SAM then serves as a methyl donor in methyltransferase reactions. A necessary byproduct of this process is S-adenosyl homocysteine (SAH) [54], which is hydrolyzed by the enzyme SAHH, resulting in the formation of homocysteine (Hcy). Hcy is eliminated from circulation either through conversion to homocysteine thiolactone or through the trans-sulfuration pathway, leading to the formation of cysteine. Alternatively, remethylation can occur through two pathways: one pathway utilizes methionine synthase with B12 as a cofactor and 5-methyltetrahydrofolate as the methyl donor, and the other involves betaine homocysteine methyltransferase with betaine as the methyl donor. Overall, the Met cycle connects and interacts with three vital metabolic pathways by supplying substrates (Figure 1).
2. Met与蛋氨酸循环的关系
Met 是一种必需氨基酸,其代谢受称为蛋氨酸循环的核心循环控制。在这个循环中,Met通过Met腺苷转移酶(MAT)的作用转化为SAM。然后,SAM在甲基转移酶反应中充当甲基供体。该过程的必要副产物是S-腺苷同型半胱氨酸(SAH)[54],它被SAHH酶水解,形成同型半胱氨酸(Hcy)。Hcy通过转化为同型半胱氨酸硫内酯或通过反式硫化途径从循环中消除,导致半胱氨酸的形成。或者,再甲基化可以通过两种途径发生:一种途径利用以 B12 为辅因子和 5-甲基四氢叶酸作为甲基供体的蛋氨酸合酶,另一种途径涉及以甜菜碱为甲基供体的甜菜碱同型半胱氨酸甲基转移酶。总体而言,Met循环通过提供底物与三种重要的代谢途径连接并相互作用(图Figure 1)。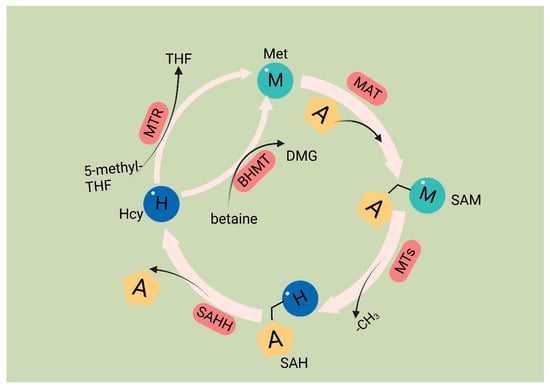
Methionine cycle.The first step of the methionine cycle is the conversion of methionine into S-adenosylmethionine (SAM) by methionine adenosyltransferase (MAT). SAM is converted into S-adenosyl homocysteine (SAH) after donating a methyl group for methylation reactions. This step is mediated by methyltransferases (MTs). SAH is then hydrolyzed by S-adenosyl-L-homocysteine hydrolase (SAHH) to generate homocysteine. Finally, homocysteine receives a methyl group from the folate cycle or betaine to become methionine; these reactions are mediated by 5-methyltetrahydrofolate: homocysteine methyltransferase (MTR) and betaine-homocysteine methyltransferase (BHMT), respectively. The methionine cycle is interconnected with three important metabolic pathways by providing substrates. These pathways include the folate cycle, the transsulfuration pathway, the methionine salvage pathway, and polyamine synthesis, all of which support important cellular functions. Met: methionine; SAM: S-adenosyl methionine; MAT: Met adenosyltransferase; SAH: S-adenosylhomocysteine; MTs: methyltransferases; Hcy: homocysteine; DMG: dimethylglycine; BHMT: betaine-homocysteine methyltransferase; 5-methyl-THF: 5-methyltetrahydrofolate; THF: tetrahydrofolate; MTR: methyltransferase.
In cancer cells, the deficiency of Met leads to the abnormal recognition of translation initiation sites, thereby inhibiting protein synthesis [55]. The significance of Met in cancer cell function has been extensively studied. Metabolites generated through the metabolic cycle play crucial roles in various intracellular processes, including polyamine synthesis, DNA synthesis, redox homeostasis, and methylation reactions [19]. SAM serves as a methyl donor for methyltransferases (MTases), enzymes that facilitate the transfer of methyl groups to various biomacromolecules, including DNA, RNA, proteins, and other metabolites that require methylation through the addition of methyl groups [56]. After losing the methyl group, SAM is converted to SAH. SAH has a significant impact on the catalytic ability of DNA methyltransferases (DNMTs) and histone methyltransferases (HMTs) [57]. SAH determines the activity of DNA and histone methyltransferases [54]. SAH undergoes hydrolysis to produce Hcy. Hcy has two potential fates; it can either enter the transsulfuration pathway or be methylated to form Met, thus connecting the Met cycle to the transsulfuration pathway. The entry of Hcy into the transsulfuration pathway leads to the production of glutathione (GSH), which plays a role in redox homeostasis. Reactive oxygen species (ROS) present in cancer cells can promote cancer cell proliferation through the PI3K pathway [58]. GSH functions as an antioxidant by reacting with ROS, scavenging them, and forming oxidized glutathione (GSSG) on its own [59].
- Mechanisms of Met in the Pathophysiology of Gastric Cancer
The reliance on Met is evident in the gastric cancer cell line, which implies that leveraging this biochemical disparity between normal and malignant cells may have potential therapeutic implications [60].
Sufficient levels of Met are essential for human cells. In the event of inadequate exogenous methionine intake to sustain regular physiological processes, normal human cells can internally utilize the residual Hcy to synthesize Met [61]. Various studies have demonstrated that tumor cells experience a heightened demand for Met due to their rapid proliferation. However, tumor cells are incapable of utilizing Hcy to synthesize Met for several reasons, with the leading cause being the ineffectiveness of the Met synthase enzyme in cancer cells. Thus, tumor cells are more sensitive to the MR environment [60,62–64]. Nevertheless, our comprehension of the role of Met metabolism in cancer remains at an early stage due to its intricate nature. On a positive note, there has been a substantial increase in published studies in recent years, shedding more light on the impact of Met on gastric cancer.
3.1. Mechanisms of Met’s Role in Helicobacter pylori Survival and Infection
Helicobacter pylori (Hp) infection is a significant risk factor for gastric cancer and is classified as a Group 1 carcinogen by the International Agency for Research on Cancer (IARC) [4]. Following infection with Hp, the adjacent gastric epithelial cells undergo a complex inflammatory response. This environment triggers the release of various cytokines, reactive oxygen species (ROS), and nitric oxide (NO) by immune cells. These inflammatory mediators subsequently activate DNA methyltransferases, resulting in the hypermethylation of CpG islands and ultimately leading to the downregulation of associated gene expression [65]. Furthermore, gastric epithelial cells undergo epigenetic alterations following Hp infection, including the methylation of E-calmodulin, a process that may contribute to gastric cancer metastasis [66]. Epigenetic alterations significantly improve after Hp eradication, leading to a delay in the development of Hp-induced gastric cancer [67]. Hp faces multiple challenges to thrive in the stomach, such as the acidic environment and oxidative stress [68–70]. Hp possesses multiple mechanisms to counteract oxidative stress. One prominent mechanism involves the enzyme catalase, which facilitates the conversion of hydrogen peroxide and oxygen into less detrimental compounds. Prior research indicates that Met residues in peroxidases and ureases can serve a non-catalytic function to mitigate oxidative stress. The specific mechanism involves the binding of Met residues to oxidizing agents, resulting in the formation of methionine sulfoxide (Met-SO), which can subsequently be restored to methionine residues by methionine sulfoxide reductase (Msr). This establishes a Met-S/Met-SO cycle, effectively reducing oxidative damage in Hp [49,71].
Moreover, evidence shows that Hcy, a direct precursor of Met, synthesizes endogenous hydrogen sulfide (H2S) through a degradation pathway. Hcy is converted to H2S via three pathways: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfotransferase (3MST) [72]. This metabolic process promotes vasodilation angiogenesis, inhibits leukocyte adhesion to vessel walls, and upregulates antioxidant molecules [73,74]. Some studies have suggested that H2S is a protective factor, mitigating chronic inflammation, including gastric mucosal atrophy induced by Hp [75]. H2S is a reducing agent that scavenges oxidizing molecules, including ROS and hydrogen peroxide. Additionally, H2S enhances wound healing by facilitating angiogenesis through the phosphatidylinositol 3-kinase/Akt signaling pathway [76–78]. Furthermore, H2S restricts neutrophil migration, inflammation, and oxidative burst [79] (Figure 2).
Figure 12.
3. Met在胃癌病理生理学中的机制
3.1. Met在幽门螺杆菌存活和感染中的作用机制
幽门螺杆菌(Helicobacter pylori, Hp)感染是胃癌的重要危险因素,被国际癌症研究机构(International Agency for Research on Cancer, IARC)列为1类致癌物[4]。感染 Hp 后,邻近的胃上皮细胞会经历复杂的炎症反应。这种环境触发免疫细胞释放各种细胞因子、活性氧 (ROS) 和一氧化氮 (NO)。这些炎症介质随后激活DNA甲基转移酶,导致CpG岛的高甲基化,并最终导致相关基因表达的下调[65]。此外,Hp感染后,胃上皮细胞会发生表观遗传改变,包括E-钙调蛋白的甲基化,这一过程可能导致胃癌转移[66]。根除Hp后,表观遗传改变显著改善,导致Hp诱导的胃癌发生延迟[67]。Hp在胃中茁壮成长面临多种挑战,例如酸性环境和氧化应激[68,69,70]。Hp 具有多种抵消氧化应激的机制。一个突出的机制涉及过氧化氢酶,它促进过氧化氢和氧气转化为危害较小的化合物。先前的研究表明,过氧化物酶和脲酶中的Met残基可以起到非催化作用,以减轻氧化应激。具体机理涉及Met残基与氧化剂结合,导致蛋氨酸亚砜(Met-SO)的形成,随后可通过蛋氨酸亚砜还原酶(Msr)还原为蛋氨酸残基。这建立了Met-S/Met-SO循环,有效减少了Hp中的氧化损伤[49,71]。 此外,有证据表明,Met的直接前体Hcy合成内源性硫化氢(HMet plays a pivotal role in the survival and functional repertoire of Hp. Hp infection destroys the mucus layer of the stomach, leading to chronic inflammation within the stomach. During this period, Hp is damaged by both the acidic environment within the stomach and oxidizing factors secreted by inflammatory cells. Hp is able to resist the acidic environment through urease. And it utilizes Met to counteract the antioxidant factors. The S (Met-S) of the Met residue of urease and catalase reacts with oxidizing factors to convert high oxidizing factors to gastric low oxidizing factors, converts itself to Met-SO, and then converts back to Met-S when catalyzed by methionine sulfoxide reductase. Hcy is converted to H2S via three pathways: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfotransferase (3MST). H2S is a reducing agent that scavenges oxidizing molecules, including ROS and hydrogen peroxide (H2O2). Additionally, H2S enhances wound healing by facilitating angiogenesis through the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway. Furthermore, H
S restricts neutrophil migration, inflammation, and oxidative burst.
In a Met-rich environment, Met provides aid in Hp’s defense against oxidative stress, thus promoting Hp survival and activity. Conversely, Hcy-rich environments generate H
3.2. Met在胃癌细胞程序性细胞死亡中的机制
程序性细胞死亡,称为细胞凋亡,是一种由遗传决定的、活跃的、有序的细胞死亡过程。这种机制起到了防止内部和外部环境因素刺激的保护措施的作用。有四种公认的程序性细胞死亡类型:细胞凋亡、程序性坏死、细胞焦亡和铁死亡。 此外,MR环境中的胃癌细胞没有表现出腹膜扩散,并且在体内实验中观察到了相同的有利结果。后续研究表明,MR 培养基完全去甲基化了内源性 E-cadherin (CDH1) 基因,并增强了 MKN45 和 KATOIII 细胞系中 CDH1 的表达。CDH1 是一种细胞间粘附蛋白。在弥漫性胃癌病例中,CDH1的启动子区通常被高甲基化,这与CDH1蛋白表达的下调密切相关;此外,临床证据表明,CDH1蛋白表达的下调会导致胃癌表型更具侵袭性,胃癌患者的预后更差[81,82]。 最近的研究表明[46],Met裂解酶(Metase)的过表达显著促进胃癌细胞的凋亡和自噬。此外,用Metase转染SGC7901和MKN45细胞系可显著增强细胞凋亡。值得注意的是,Beclin1、Atg5 和 Atg7 蛋白的表达以及自噬标志物 LC3-I 比率显着升高。 研究[45]发现,重组蛋氨酸酶(rMetase)处理后,胃癌细胞系中PI1K的表达、磷酸化Akt(p-Akt)与总Akt(t-Akt)的比值、GLUT-2和关键糖酵解酶(如HK2、PFKM和LDHA)的表达下调。此外,抗凋亡蛋白 Bcl-3 下调,而促凋亡蛋白 Bax 和半胱氨酰天冬酰胺-7901 上调。临床证据表明,BCL-3下调可延长胃癌患者的生存期[86]。 铁死亡是一种独特的非凋亡细胞死亡调节形式,主要由脂质过氧化过度触发[90]。它是最近发现的一种程序性细胞死亡机制,主要在肿瘤细胞中观察到。代谢重编程有可能改变对铁诱导的细胞死亡的敏感性。细胞中铁、脂质和谷氨酰胺的代谢被认为是影响细胞对铁中毒易感性的关键代谢过程[90,91]。众所周知,细胞受到三个主要抗氧化轴的保护,即胱抑素/GSH/GPX4轴、FSP1/CoQ10轴和GCH1/BH4/DHFR轴[92]。3.3. Met在胃癌免疫反应中的作用机制
由免疫系统引起的慢性炎症被广泛认为是癌症发展的一个突出因素[95]。因此,癌症被某些学者称为“持续性伤口”[96]。毫无疑问,免疫系统在癌症的发展和进展中起着至关重要的作用[97]。一个说明性的例子是幽门螺杆菌诱发慢性胃炎的能力,从而将幽门螺杆菌确定为胃癌发展的重要危险因素。此外,在癌症晚期,免疫系统可以刺激癌细胞迁移并增强恶性细胞的侵袭性[98]。免疫系统对癌症的影响延伸到肿瘤微环境。癌细胞通过诱导免疫细胞因子释放来利用免疫细胞,导致微环境改变,促进肿瘤血管生成和组织重塑[99,100]。由于癌细胞和宿主细胞之间的异质性以及癌细胞的快速生长,癌细胞与免疫系统有关。当癌细胞的生长速度超过血液中的能量供应时,就会发生坏死,导致细胞碎片的产生和大量的促炎因子。这些因子激活免疫系统,随后募集免疫细胞[101,102]。 临床和实验室证据表明,癌细胞能够通过吸收环境中的大量Met来逃避免疫系统[105,106,107]。MR疗法有可能被用作抑制癌症进展的免疫疗法,这并不意外[108]。 已经证明,MR 或抑制 MATA2 通过抑制组蛋白 H3 (H3K4me3) 上的赖氨酸-4 和组蛋白 H3 (H3K27me3) 上的赖氨酸-27 的三甲基化来降低 RIP1 的表达。因此,这种抑制会阻碍胃癌中单核细胞/巨噬细胞的浸润[109]。单核细胞/巨噬细胞是胃癌免疫细胞浸润的重要组成部分[110]。 总体而言,Met在胃癌免疫中的作用至关重要。MR不仅抑制胃癌细胞的发育,还阻碍单核细胞/巨噬细胞的浸润。虽然补充Hcy对MR对胃癌细胞的影响不受影响,但它减轻了癌细胞对T细胞的竞争性抑制,促进了T细胞的增殖和分化。虽然胃癌细胞表现出异质性,但体内的免疫细胞是我们的正常细胞,不同胃癌患者之间同类型的免疫细胞差异很小。因此,受Met影响的机制被认为是可推广的。3.4. MET 对胃癌干细胞影响的机制
胃癌干细胞 (GCSC) 是一种特定的癌细胞亚型,其特征在于它们能够自我更新并分化为其他癌细胞类型。人们普遍认为,在胃癌中观察到的侵袭、转移和对化疗的耐药性可归因于 GCSC 的存在和活性。这些细胞在胃癌的进展和治疗反应中起重要作用[119]。由于GCSC的快速扩散速度,对Met的需求很高。在喂食MR但暴露于Hcy的小鼠中,GCSCs的增殖率显着降低。RAB37 是一种小的 GTP 酶蛋白,负责细胞信号传导和囊泡转运,可通过与自噬相关基因 5 (ATG5) 结合诱导自噬;此外,临床研究表明,ATG5下调促进胃癌侵袭,RAB37表达下调与癌症转移和预后不良有关[120,121,122]。后续研究表明,MR通过两种途径增强了RAB37的作用,促进了GCSCs的自噬,并降低了GCSCs的增殖率。 首先,MR降低了RAB37的甲基化,导致RAB37表达的上调。此外,调节 miR-200b/PKCα 轴间接增加了 RAB37 的活性。这些作用导致GCSCs的自噬增加和增殖率降低[40]。3.5. MET在胃癌化疗中的作用和机制
尽管在胃癌病例中观察到显着的异质性,但晚期患者的治疗方法差异很小。通常,对于局限性晚期癌症患者,标准治疗包括切除后辅助化疗[128]。辅助化疗通常需要一种称为 FLOT 的方案,该方案由多西紫杉醇、奥沙利铂、5-氟尿嘧啶和亚叶酸组成。这种围手术期化疗药物的组合已成为西方国家的标准治疗[129]。对于晚期弥漫性胃癌患者,对于被认为不适合手术的患者,应考虑选择新辅助放疗[130]。 Met是甲基供体的体内来源。MR诱导被困在G0/G1期的癌细胞进入S/G2期,随后停滞[134,135]。这是细胞DNA经历快速复制的时期,使其成为癌细胞对化疗高度敏感的黄金时间。因此,从理论上讲,MR可以增强化疗药物治疗癌症的疗效。越来越多的研究表明,MR可以与各种化疗药物协同作用;例如,大都会的癌细胞−/Hcy+培养基选择性地阻滞在S/G2期,进一步消除克隆细胞,使癌细胞对细胞周期特异性药物敏感[64],包括5-氟尿嘧啶[136]、吉西他滨[137]、顺铂[52]、多柔比星(DOX)、长春新碱(VCR)[138]等。除了诱导化疗敏感的 S/G2 期外,MR 还可能通过替代途径增强化疗药物的疗效。由于通过定期喂养实现 MR 的挑战,研究人员利用易于获得的蛋氨酸酶 (METase) 作为创建 MR 环境的更直接方法。一项研究表明[52]METase能够恢复耐药胃癌细胞对顺铂的敏感性。其具体机制涉及抑制METase治疗后胃癌细胞中核因子-κB(NF-κB)活性,导致肿瘤坏死因子相关凋亡诱导配体(TRAIL)表达上调,随后P-gp下调。此外,NF-κB的失活导致miR-21表达下调,进一步增强了胃癌细胞对顺铂的敏感性[139]。NF-κB是一种转录因子,可以激活抗凋亡蛋白等分子,从而减少胃癌细胞的凋亡[140]。TRAIL可以选择性地诱导癌细胞死亡,而不会对正常细胞造成伤害[141](图S, proving to be detrimental to Hp’s survival and activity. Theoretically, an MR strategy involving low Met and high Hcy could potentially delay gastric carcinogenesis by suppressing Hp survival and activity. The entire process does not directly involve gastric cancer cells; therefore, theoretically, the heterogeneity of gastric cancer cells would not affect the effectiveness of MR.
3.2. The Mechanism of Met in Programmed Cell Death of Gastric Cancer Cells
Programmed cell death, known as apoptosis, is a genetically determined, active, and orderly process through which cells die. This mechanism functions as a protective measure against stimuli from both internal and external environmental factors. There are four recognized types of programmed cell death: apoptosis, programmed necrosis, cellular pyroptosis, and ferroptosis. A study conducted in 2013 [80] observed that MR induces apoptosis and inhibits cell adhesion and migration in gastric cancer cells. The number of cancer cells cultured in the MR medium significantly reduced compared to the control group.
Furthermore, gastric cancer cells within the MR environment did not exhibit peritoneal spreading, and the same favorable outcome was observed in the in vivo experiments. Subsequent studies demonstrated that the MR medium fully demethylated the endogenous E-cadherin (CDH1) gene and enhanced CDH1 expression in the MKN45 and KATOIII cell lines. CDH1 is an intercellular adhesion protein. The promoter region of CDH1 is commonly hypermethylated in cases of diffuse gastric cancer, which is closely linked to the downregulation of CDH1 protein expression; moreover, clinical evidence has demonstrated that the downregulation of CDH1 protein expression leads to a more aggressive phenotype of gastric cancer and a worsened prognosis for patients with gastric cancer [81,82]. These studies demonstrated that MR induces apoptosis and suppresses the invasive metastasis of gastric cancer cells.
Recent studies [46] have indicated that the overexpression of the Met cleavage enzyme (Metase) significantly promotes apoptosis and autophagy in gastric cancer cells. Furthermore, the transfection of the SGC7901 and MKN45 cell lines with Metase substantially enhances apoptosis. Notably, the expression of Beclin1, Atg5, and Atg7 proteins and the autophagy marker LC3-I ratio were considerably elevated. Additionally, further investigations revealed that the expression of SNHG5 was reduced, while miR-20a expression was increased in Metase-transfected cancer cells, and there is clinical evidence that miR20a promotes cancer development [83]. Furthermore, the effect observed in cancer cells transfected with Metase was nullified upon adding si-SNHG. Notably, the inhibition of SNHG5 with si-SNHG5 reduced miR-20a expression, thereby promoting the proliferation of gastric cancer cells [84,85]. Thus, it can be concluded that the depletion of Met in gastric cancer cells stimulates autophagy and apoptosis, thereby hindering the proliferation of gastric cancer cells.
It was discovered [45] that the expression of PI1K, the ratio of phosphorylated Akt (p-Akt) to total Akt (t-Akt), GLUT-2, and key glycolytic enzymes, such as HK2, PFKM, and LDHA, were downregulated in gastric cancer cell lines following treatment with recombinant methioninase (rMetase). Additionally, the anti-apoptotic protein Bcl-3 was downregulated, while the pro-apoptotic proteins Bax and cysteinyl asparagin-7901 were upregulated. Clinical evidence suggests that BCL-3 downregulation prolongs survival in gastric cancer patients [86]. Recent research has demonstrated that the gastric cancer cell viability and proliferation rate decreased, and the expression of long non-coding RNA (lncRNA) PVT1 was significantly reduced after culturing in a medium lacking Met. Furthermore, further investigations showed that the interaction between lncRNA PVT1 and DNMT3 impacted the DNA methylation level of the BNIP1 promoter. This interaction, along with the downregulation of lncRNA PVT1 and the upregulation of BNIP3 levels, inhibited the proliferation of gastric cancer cells, and clinical evidence demonstrates that BNIP3 upregulation favors the prognosis of cancer patients [87,88]. The deprivation of Met can upregulate the expression of BNIP1 by inhibiting the binding between lncRNA PVT3 and DNMT1, which, in turn, activates mitochondrial autophagy and ultimately inhibits the proliferation of gastric cancer cells [89].
Ferroptosis is a distinct form of non-apoptotic cell death regulation primarily triggered by excessive lipid peroxidation [90]. It is a recently identified programmed cell death mechanism mainly observed in tumor cells. Metabolic reprogramming has the potential to alter the sensitivity to iron-induced cell death. The metabolism of iron, lipids, and glutamine in cells is recognized as a crucial metabolic process that influences the vulnerability of cells to iron poisoning [90,91]. It is well known that cells are protected from iron oxidation by three principal antioxidant axes, i.e., the cystatin/GSH/GPX4 axis, FSP1/CoQ10 axis, and GCH1/BH4/DHFR axis [92]. Recent studies have shown that the inhibition of MR or the SAM-generating enzyme Met adenylyltransferase 2A (MAT2A) results in gastric cancer cells that are more susceptible to iron apoptosis. Ferroptosis inhibitors can eliminate this effect. Further studies revealed that MR-induced ferroptosis is not caused by cell cycle disruption or decreased cell viability, but it is a direct result of Met cycle disruption. The pharmacological inhibition of MAT2A resulted in a significant decrease in histone H3K4me3 trimethylation in the ACSL3 promoter region (acyl-coenzyme A synthetase long-chain family member 3), leading to the downregulation of ACSL3 expression [55]. ACSL3 plays a pivotal role as the central enzyme in the process of generating fatty acyl-coenzyme A esters. Moreover, incorporating these esters into membrane phospholipids provides a protective mechanism for cells against ferroptosis-induced mortality [93]. Thus, the activation of the MR receptor or the inhibition of the MATA2 protein promotes the occurrence of ferroptosis in gastric cancer cells. This promotion is achieved by suppressing the trimethylation of histone H3 at lysine 4 (H3K4me3), reducing the expression levels of ACSL3, and clinical evidence demonstrates that ACSL3 downregulation benefits the prognosis of cancer patients [44,94] (Figure 3).
Figure 3. MR promotes the programmed cell death of gastric cancer cells. In the MR environment, the CDH1 promoter was completely demethylated, upregulating the expression of CDH1 and thus enhancing the expression of E-cadherin and inhibiting the invasive metastasis of gastric cancer. The downregulation of SNHG5 expression and the increase in miR-20a expression promoted the autophagy and apoptosis of gastric cancer cells; the expression of long non-coding RNA (lncRNA) PVT1 was also significantly downregulated, and the interaction between lncRNA PVT1 and DNMT3 was weakened. The expression of long non-coding RNA (lncRNA) PVT1 was also significantly downregulated, and the interaction between lncRNA PVT1, and DNMT3 was weakened, leading to the decrease in the DNA methylation level of the BNIP1 promoter and the upregulation of the level of BNIP3, which suppressed the proliferation of gastric cancer cells and activated the autophagy of mitochondria, leading to the apoptosis of gastric cancer cells.
When reviewing these studies, it becomes apparent that Met, being a critical methyl source, can modulate apoptosis, autophagy, and ferroptosis in gastric cancer cells by regulating genetic material and protein methylation. This effect is not limited to any specific molecule. Therefore, despite the heterogeneity observed among individual gastric cancer cells and even among gastric cancer cells within the same entity, it is theoretically improbable for MR to promote programmed cell death in gastric cancer cells.
3.3. The Mechanism of Met’s Role in the Immune Response to Gastric Cancer
Chronic inflammation, caused by the immune system, is widely recognized as a prominent factor in cancer development [95]. Consequently, cancer has been dubbed by certain scholars as “the persistent wound” [96]. Undoubtedly, the immune system plays a crucial role in the development and progression of cancer [97]. An illustrative example is the ability of Helicobacter pylori to induce chronic gastritis, thereby establishing H. pylori as a significant risk factor for the development of gastric cancer. Moreover, during the advanced stages of cancer, the immune system can stimulate cancer cell migration and augment the invasiveness of malignant cells [98]. The impact of the immune system on cancer extends to the tumor microenvironment. Cancer cells employ immune cells by inducing their cytokine release, leading to microenvironmental alterations that facilitate tumor angiogenesis and tissue remodeling [99,100]. Cancer cells are associated with the immune system due to both the heterogeneity between cancer cells and host cells and the rapid growth of cancer cells. When cancer cells grow at a pace that exceeds the energy supply in the bloodstream, necrosis occurs, resulting in the production of cellular debris and an abundance of pro-inflammatory factors. These factors activate the immune system, subsequently recruiting immune cells [101,102]. Moreover, it facilitates cancer development, entangled in a self-perpetuating loop. Nonetheless, the immune system also plays a role in suppressing cancer [103]. Upon exposure to tumor antigens, T cells become activated and differentiate into cytotoxic effector T cells, which are proficient in eliminating cancer cells. Consequently, T cells play a pivotal role in cancer immunotherapy [104].
Clinical and laboratory evidence suggests that cancer cells are capable of evading the immune system by assimilating significant quantities of Met from the environment [105–107]. It is not unexpected that MR therapy might have the potential to be utilized as an immunotherapy to restrain cancer progression [108].
It has been demonstrated that MR or inhibition of MATA2 reduce RIP1 expression by suppressing the trimethylation of lysine-4 on histone H3 (H3K4me3) and lysine-27 on histone H3 (H3K27me3) at the promoter of RIP1. As a result, this inhibition hinders the infiltration of monocytes/macrophages in gastric cancer [109]. Monocytes/macrophages are a significant component in the immune cell infiltration of gastric cancer [110]. Similarly, methionine exerts a substantial influence on T cell activity [111–113]. Upon antigen recognition, T cells undergo rapid proliferation and differentiation, requiring a substantial methyl supply from Met for efficient completion of DNA methylation and histone methylation [114,115]. Furthermore, studies have demonstrated that when T cells are stimulated in the absence of Met, there is a decrease in histone H3K4me3 modification. This reduction in histone methylation subsequently inhibits both T cell proliferation and differentiation [116]. In mouse Th17 cells, the activation of MR results in a significant reduction in the secretion of interleukin-17 (IL-17) and interferon-gamma (IFN-γ), thereby compromising the functionality of T cells. This has important implications in cancer tissues, as cancer cells heavily uptake the surrounding Met, thereby depriving T cells of an adequate supply of this essential nutrient. Consequently, the insufficiency of Met acquisition by T cells enables cancer cells to evade T cell-mediated elimination, thereby promoting their survival and progression [105]. In the earlier discussion, it was noted that normal cells can utilize homocysteine to synthesize methionine for their essential cellular functions, while cancer cells lack this capability. Consequently, by supplementing an adequate amount of homocysteine during MR in cancer cells, it is possible to hinder the proliferation of cancer cells and reactivate T cells to eliminate cancer cells. There have been previous studies showcasing the promotion of T cell proliferation and differentiation by Hcy. However, a research gap exists in terms of additional investigation [117,118].
Overall, the role of Met in gastric cancer immunization is crucial. MR not only inhibits the development of gastric cancer cells but also hampers the infiltration of monocytes/macrophages. Although the impact of Hcy supplementation on the effect of MR on gastric cancer cells remains unaffected, it alleviates the competitive inhibition of T cells caused by cancer cells and promotes T cell proliferation and differentiation. While gastric cancer cells display heterogeneity, immune cells in the body are our normal cells, and there is minimal variation in the same type of immune cells among different gastric cancer patients. Thus, the mechanisms influenced by Met are presumed to be generalizable.
3.4. Mechanisms Underlying the Impact of Met on Gastric Cancer Stem Cells
Gastric cancer stem cells (GCSCs) are a specific subtype of cancer cells characterized by their ability to self-renew and differentiate into other cancer cell types. It is widely acknowledged that the invasion, metastasis, and resistance to chemotherapy observed in gastric cancer can be attributed to the presence and activities of GCSCs. These cells play significant roles in the progression and treatment response of gastric cancer [119]. Due to the fast proliferation rate of GCSCs, the demand for Met is high. The proliferation rate of GCSCs was significantly reduced in mice fed with MR but exposed to Hcy. RAB37, a small GTPase protein, is responsible for cellular signaling and vesicular transport and can induce autophagy by binding to autophagy-related gene 5 (ATG5); in addition, clinical studies have shown that ATG5 downregulation promotes gastric cancer invasion, and the downregulation of RAB37 expression is associated with cancer metastasis and a poor prognosis [120–122]. Subsequent studies revealed that MR enhanced RAB37’s action through two pathways, promoting autophagy in GCSCs and resulting in a decrease in the proliferation rate of GCSCs. Firstly, MR decreased the methylation of RAB37, resulting in the upregulation of RAB37 expression. Additionally, regulating the miR-200b/PKCα axis indirectly increased the activity of RAB37. These actions led to increased autophagy and a decreased proliferation rate of GCSCs [40]. Additionally, Met can regulate the methylation of non-coding RNAs, thus affecting the proliferation of GCSCs. Various studies have demonstrated that miR-7, a non-coding RNA, functions as a tumor suppressor in different cancer types, including gastric cancer. Specifically, miR-7 suppresses the invasion and metastasis of gastric cancer through the inhibition of the epidermal growth factor receptor (EGFR) and nuclear factor-κB (NF-κB) signaling pathway [123,124]. There are also relevant clinical data that show that miR-7 inhibits gastric cancer proliferation [125]. Nevertheless, the expression of miR-7 in GCSCs was found to be suppressed due to an increase in DNA methylation of the miR-7 promoter in GCSCs. GCSCs cultured in an MR medium exhibited elevated miR-7-5p expression, which correlated with a reduced invasiveness of GCSCs. Subsequent studies revealed that MR treatment activates p53 signaling in GCSCs, selectively triggers apoptosis [126], and causes a decrease in the DNA methylation level of the miR-7-5p promoter region, thereby further promoting apoptosis in GCSCs [127] (Figure 4).
Figure 4. MR suppresses gastric cancer development by inhibiting GSCSs. MR decreases the methylation of RAB37, resulting in the upregulation of RAB37 expression. Additionally, regulating the miR-200b/PKCα axis indirectly increased the activity of RAB37; then, binding it to autophagy-related gene 5 (ATG5) induced the autophagy of GCSCs. MR decreases the methylation of miR-7, resulting in the upregulation of miR7 expression, leading to the inhibition of epidermal growth factor receptor (EGFR) and nuclear factor-κB (NF-κB) signaling pathways, eventually inhibiting gastric cancer invasion and metastasis. MR activates p53 signaling in GCSCs, selectively triggers apoptosis, and causes a decrease in the DNA methylation level of the miR-7-5p promoter region, thereby further promoting apoptosis in GCSCs.
Multiple studies have demonstrated that MR can effectively inhibit the proliferation of GCSCs through various mechanisms. These findings indicate that MR has a profound impact on gastric cancer, and, importantly, this impact is not quickly diminished by the heterogeneity observed among gastric cancer cells.
3.5. The Role and Mechanism of Met in Gastric Cancer Chemotherapy
Despite the significant heterogeneity observed among cases of gastric cancer, there is minimal variation in the treatment approach for patients with advanced stages of the disease. Typically, for patients with limited advanced-stage cancer, the standard treatment involves resection followed by adjuvant chemotherapy [128]. Adjuvant chemotherapy commonly entails a regimen known as FLOT, which consists of docetaxel, oxaliplatin, 5-fluorouracil, and folinic acid. This combination of perioperative chemotherapeutic agents has become the standard of care in Western countries [129]. In cases of advanced diffuse gastric cancer, the option of neoadjuvant radiotherapy should be considered for patients who are deemed unsuitable for surgery [130]. In China, the majority of patients with gastric cancer who seek medical attention are already in advanced stages of the disease [131]. The effectiveness of chemotherapeutic drugs relies on tumor cells being in an active replication phase, which is essential for their action. However, in practice, only a small proportion of tumor cells are actively replicating, while the majority of cancer cells are in a quiescent state known as the G0/G1 phase. This quiescent state poses a significant challenge to the efficacy of chemotherapeutic drugs [132]. Moreover, cancer cells in the G0/G1 stage are more prone to migration compared to cells in other stages [133].
Met functions as an in vivo source of methyl donors. MR induces the entry of cancer cells, which are trapped in the G0/G1 phase, into the S/G2 phase, where they subsequently stagnate [134,135]. This is the period during which cellular DNA undergoes rapid replication, making it the prime time for cancer cells to be highly susceptible to chemotherapy. Thus, in theory, MR could augment the efficacy of chemotherapeutic drugs for treating cancer. A growing body of research has demonstrated that MR can synergistically interact with various chemotherapeutic agents; for instance, cancer cells in Met-/Hcy+ medium selectively arrested in the S/G2 phase and furthermore eliminated clonogenic cells and rendered cancer cells sensitive to cell-cycle-specific drugs [64], including 5-fluorouracil [136], gemcitabine [137], cisplatinum [52], Doxorubicin (DOX), Vincristine (VCR) [138], and so on. In addition to inducing the chemo-sensitive S/G2 phase, MR has the potential to augment the efficacy of chemotherapeutic agents through alternative pathways. Due to the challenge of achieving MR through regular feeding, researchers have utilized the easily obtainable methioninase (METase) as a more straightforward means of creating the MR environment. One study demonstrated [52] the ability of METase to restore sensitivity to Cisplatin in drug-resistant gastric cancer cells. The specific mechanism involves the inhibition of nuclear factor-κB (NF-κB) activity in gastric cancer cells after treatment with METase, resulting in the upregulation of tumor-necrosis-factor-associated apoptosis-inducing ligand (TRAIL) expression and the subsequent downregulation of P-gp. Furthermore, the inactivation of NF-κB results in the downregulation of miR-21 expression, further enhancing the sensitivity of gastric cancer cells to Cisplatin [139]. NF-κB, a transcription factor, can activate molecules such as anti-apoptotic proteins, thus diminishing apoptosis in gastric cancer cells [140]. TRAIL can selectively induce cancer cell death without causing harm to normal cells [141] (Figure 5).
Figure 5
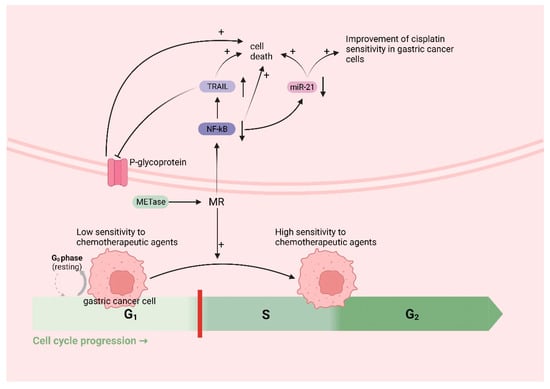
MR improves chemotherapy efficacy in gastric cancer: MR induces the entry of cancer cells, which are trapped in the G0/G1 phase (low sensitivity to chemotherapeutic agents), into the S/G2 phase (high sensitivity to chemotherapeutic agents). MR downregulates nuclear factor-κB (NF-κB), which leads to the upregulation of tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) expression, followed by the downregulation of P-gp and the death of cancer cells; the downregulation of miR-21 expression, which further enhances the sensitivity of gastric cancer cells to cisplatin and induces the death of cancer cells; and the downregulation of NF-κB, inducing the death of cancer cells.
Furthermore, growing clinical evidence suggests MR is a promising new cancer treatment modality [64]. A late phase II randomized multicenter trial involving 138 individuals with advanced gastric cancer showed that treatment with MR total parenteral nutrition for 14 days along with 5-fluorouracil and mito-mycin C resulted in a higher therapeutic response (26.3%) compared to the control group, which was given conventional Met-containing total parenteral nutrition with 5-fluorouracil and mitomycin C (8.1%). The difference was found to be statistically significant [27]. The safety of MR therapy has also been demonstrated. The consecutive brief period of a methionine-restricted (MR) diet combined with chemotherapy did not affect the patients’ nutritional statuses, evidenced by stable body weights and consistently constant plasma albumin and prealbumin levels. The combination also displayed acceptable toxicity, mainly affecting hematological parameters, particularly the platelet count [142]. In addition, a recent case report [143] showed that a 62-year-old female patient with breast cancer experienced a recurrence of metastasis in the axillary lymph nodes after four years of treatment. Due to the localized nature of the metastasis in the axillary lymph nodes, the treatment plan involved administering neoadjuvant chemotherapy comprising 3 months of doxorubicin and cyclophosphamide, followed by 3 months of docetaxel. Additionally, during the 6 months of chemotherapy, the patient underwent MR therapy, involving a low methionine diet and oral rMETase supplementation. The patient did not experience rMETase-related side effects throughout this period, aside from mild adverse effects resulting from chemotherapy.
The evidence presented supports the crucial role of Met in chemotherapy for gastric cancer. Furthermore, MR can enhance the sensitivity of gastric cancer cells to various chemotherapeutic agents by regulating the cell cycle. In addition, MR can enhance the efficacy of chemotherapy through multiple mechanisms. These findings suggest that MR can improve the chemosensitivity and effectiveness of gastric cancer cells without being affected by the heterogeneity of the disease.
- Conclusions and Prospects
The prognosis of advanced gastric cancer remains extremely poor, primarily due to the heterogeneity of gastric cancer cells and the monotony of gastric cancer treatment. The intra-tumoral heterogeneity and inter-tumoral heterogeneity of gastric cancer cells result in suboptimal therapeutic responses to many molecular targeted drugs, with some failing in clinical phase II and phase III trials. Even the targeted drugs currently used in treatment are essentially not suitable as first-line therapies and exhibit unstable efficacy. Therefore, molecular targeted therapy may not be the optimal choice for treating gastric cancer. Met is an essential amino acid for gastric cancer cells and plays a crucial role in the initiation and progression of gastric cancer. Additionally, the methionine cycle, centered around Met, also plays a significant role in gastric cancer. Consequently, limiting the acquisition of Met by gastric cancer cells significantly impacts the proliferation and invasion of gastric cancer. As discussed earlier, creating an MR environment can influence gastric cancer in multiple ways, independent of the heterogeneity of gastric cancer cells.
Based on the recent literature on Met and gastric cancer, we have found that Met’s influence on gastric cancer cells spans the entire process of gastric cancer development. The existing research suggests that, in the initiation stage, the presence of Met favors the survival of Hp and its impact on surrounding normal tissues. However, creating an MR environment through dietary interventions or enzyme addition makes Hp more susceptible to oxidative-stress-induced damage and impairs its survival, as well as mitigates the inflammation caused by Hp, thereby delaying the onset of gastric cancer. Regarding programmed cell death in gastric cancer cells, an MR environment can facilitate various types of programmed cell death, including autophagy, apoptosis, and ferroptosis. In terms of the immune response in gastric cancer, the role of Met is crucial. MR can inhibit the infiltration of monocytes/macrophages, relieve the competitive suppression of cancer cells on T cells, and promote T cell proliferation and differentiation, thus facilitating the T cell-mediated killing of gastric cancer cells. For GCSCs, MR can inhibit their proliferation through multiple mechanisms, thereby suppressing the growth and invasion of gastric cancer. During the chemotherapy process for gastric cancer, MR can enhance the sensitivity of gastric cancer cells to many chemotherapeutic drugs and enhance the efficacy of chemotherapy drugs through various mechanisms. Of course, the clinical application of MR therapy needs to be explored in more clinical trials, but we consider MR to be a vital component of gastric cancer treatment that bypasses the influence of gastric cancer heterogeneity, though further exploration into the underlying mechanisms is warranted. The metabolic therapy represented by MR has the potential to become a new research direction for gastric cancer.