Identification of specific biomarkers on the surface of CAFs offers a strategic avenue for targeted radiological diagnostics and therapeutics
[59][60][61][62][63][64][59,60,61,62,63,64]. Among these biomarkers, fibroblast activation protein (FAP) has gained widespread attention for its potential in CAF identification and targeting
[65][66][67][68][69][70][71][65,66,67,68,69,70,71]. FAP, a member of the dipeptidyl peptidase 4 (DPP4) family, boasts a molecular weight of 170 kDa
[72][73][74][75][76][72,73,74,75,76]. It assumes the guise of a type II transmembrane serine protease, typically existing as a homodimer
[77][78][79][77,78,79]. Functionally, FAP exhibits dipeptidyl peptidase and endopeptidase activities
[65][80][81][82][65,80,81,82], and its significance extends to normal embryonic development and tissue modeling
[83][84][85][83,84,85]. Remarkably, FAP remains scarcely noticeable or entirely absent in normal adult tissues
[86][87][86,87]. However, it undergoes marked upregulation during processes such as wound healing, atherosclerotic plaque formation, and fibrosis
[88][89][90][91][88,89,90,91], and prominently features in over 90% of human epithelial carcinomas
[72][80][92][93][94][95][96][97][98][72,80,92,93,94,95,96,97,98].
2. Antibody-Based Radiopharmaceuticals Targeting FAP
2.1. Iodine-131-Labeled Monoclonal Antibody F19
The discovery of FAP, a type II transmembrane serine protease, can be traced back to 1986 when it was initially identified as the F19 antigen during studies involving cultured fibroblasts and the monoclonal antibody (mAb) F19
[99][100][101][117,118,119]. Subsequently, in 1994, the surface antigen expressed by F19 cells was officially named FAP
[86][102][86,120]. In 1990, Garin-Chesa et al. proposed that in the context of cancer, epithelial cancer, F19
+ fibroblasts, colloquially referred to as FAP, emerged as a consistent molecular trait of the reactive stroma. The role of mAbF19 in their identification was pivotal
[94]. Human FAP, discerned through mAbF19, became a prominent cell surface antigen
[103][121]. Due to its abundant presence within the tumor mesenchyme, FAP can serve as a target for radionuclide antibody conjugates in cancer patients
[103][104][105][121,122,123].
In 1988, Old et al. conducted a comprehensive examination of six human cell surface glycoproteins, each defined by mAb, with the intention of characterizing the surface phenotype of cultivated mesenchymal cells
[92]. Among these antibodies, mAbF19 effectively identified glycoproteins with molecular weights of 120,000 and 95,000, expressed on cultivated fibroblasts and a proportion of sarcoma cell lines, respectively. This discovery marked mAbF19 as a superior antibody for these purposes compared with other candidates, such as mAbF24, G171, G253, and K117. Another antibody, S5, exhibited expression patterns similar to mAbF19 but had limited in vivo expression
[92]. It became evident that the fibroblasts surrounding tumor cells offer effective targets for cancer immunolocalization or immunotherapy, owing to their recognition by mAbF19
[106][107][108][129,130,131].
The pioneering clinical study involving FAP-targeting radiopharmaceuticals utilized
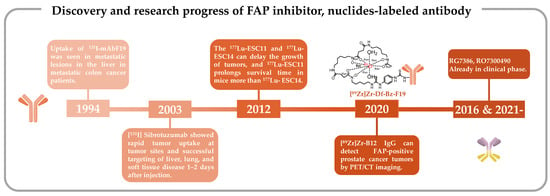
131I-labeled mAbF19 for tumor imaging in patients with liver metastases from colon cancer
[109][132]. Welt et al. conducted this study in 1994, wherein 17 patients scheduled for resection of localized metastases or regional chemotherapy received intravenous administration of
131I-mAbF19. Imaging results from this series of studies revealed that the tumor-to-normal tissue ratio reached its peak after 3–5 days of administration, enabling the visualization of lesions as small as 1 cm in diameter. Notably, colon cancer studies demonstrated specific localization of tumors and metastatic lesions. However, SPECT/CT results revealed slow kidney clearance, necessitating 3–5 days to achieve optimal imaging outcomes. This delayed renal clearance has implications for the imaging capabilities of
131I-mAbF19 (
Figure 1)
[109][132].
Figure 1. Discovery and research progress of nuclide-tagged FAP-targeting antibodies [109][110][111][112][113][114]. Discovery and research progress of nuclide-tagged FAP-targeting antibodies [126,127,128,132,133,134].
2.2. Iodine-131-Labeled Sibrotuzumab
mAbF19 stands out for its FAP-specific targeting capabilities, and scientists have endeavored to enhance its imaging potential as a radiopharmaceutical
[115][135]. A study by Welt et al. established the positive expression of FAP in all 17 patients studied, and SPECT/CT imaging effectively facilitated precise tumor identification in humans
[109][132]. This encouraged further investigation into the pharmacokinetic (PK) of sibrotuzumab, a humanized version of the murine anti-FAP mAbF19. A phase I and II clinical trial (NCT02198274) involving sibrotuzumab sought to assess its PK without radiolabeling in patients with FAP-positive malignancies, particularly advanced metastatic colorectal cancer patients. However, among the 17 patients enrolled who had undergone rigorous pretreatment, only two exhibited stable disease status, a count insufficient to meet the phase II trial criteria, which typically necessitate four patients in a stable condition or at least one patient in complete or partial remission. Consequently, the trial did not progress beyond phase II
[116][136]. The PK study unveiled pertinent findings, including a mean clearance rate of 39.8 ± 19.8 mL/h and a terminal half-life of 5.3 ± 2.3 days for sibrotuzumab.
One such avenue of exploration encompassed a clinical phase I dose-escalation study of sibrotuzumab in patients with colorectal or non-small cell lung cancer (NCT02209727). This study, conducted in parallel with the aforementioned phase I/II trials, sought to evaluate biodistribution, PKs, immunogenicity, and safety profiles by incrementally administering sibrotuzumab intravenously to 26 patients. In tandem, two antecedent phase I studies employed
131I-mAbF19, with a focus on patients with hepatic metastases from colorectal cancer or soft tissue sarcoma
[117][137]. These investigations delved into the PK parameters of the therapeutic mouse monoclonal antibody
131I-mAbF19, elucidating principles of selective tumor accumulation and stromal targeting in tumors through biodistribution imaging studies and biopsy analyses. These pivotal insights provided the foundation for the inaugural human clinical evaluation of sibrotuzumab.
In this human trial, patients received 8–10 mCi of
131I-labeled sibrotuzumab, administered concomitantly at weeks 1, 5, and 9 in a 12-week dosing cycle, with a focus on PK assessments. The ensuing analysis divulged that the mean clearance rate of
131I-sibrotuzumab amounted to 41.9 ± 16 mL/h, accompanied by a half-life of 4.9 days. Following a single cycle of
131I-sibrotuzumab treatment, two out of the 26 patients manifested stable disease conditions. The relatively abbreviated half-life of sibrotuzumab held promise for radioimmunotherapy. Regrettably, the trial did not yield definitive efficacy results for sibrotuzumab, prompting the discontinuation of further clinical development (
Figure 1)
[113][133].
2.3.
177
Lu-ESC11/ESC14
The clinical application of
131I-mAbF19 and its humanized derivative, sibrotuzumab, has been hindered by their prolonged blood clearance and suboptimal therapeutic efficacy. To enhance the therapeutic potential of FAP-targeted antibodies, it is imperative to develop antibodies with enhanced attributes and utilize radiolabeling with more suitable radionuclides
[118][119][120][121][138,139,140,141].
From the perspective of antibody discovery, ESC11 and ESC14, two antibodies noted for their selective accumulation within xenografted FAP-positive human melanoma and their capacity to impede tumor growth in vivo, were identified in the human FAP antibody library using the phage display technique. Subsequently, these antibodies underwent a transformation into IgG1 antibodies
[110][126]. The phage display technique is a new technique predicated on specific affinity interactions, enabling the identification of proteins or peptides that exhibit particular binding properties. This technique is especially adept at discovering antibodies targeting challenging and intriguing molecules, making it a cornerstone in the quest for antibodies with precise attributes
[122][123][124][125][126][142,143,144,145,146]. It enjoys widespread utilization in the quest for human antibody fragments boasting specific binding activity
[126][127][128][146,147,148]. This technique facilitates the evolution and optimization of FAP-targeting antibodies, culminating in the selection of antibodies exhibiting robust affinity, rapid internalization, and propensity for tumor accumulation.
From a radionuclide perspective, radiolabeling with iodine-131 is relatively straightforward for mAbs
[129][149]. However, in the context of radioimmunotherapy, iodine-131 proves to be suboptimal due to its propensity for facile release from tumor sites after internalization of mAbs within cells
[130][131][150,151]. A related drawback lies in the emission of high-energy (364 keV) γ-photons, accounting for a substantial 82% of its radiation output, thus posing concerns for radiation safety
[132][133][105,152]. In stark contrast, the radioactive lanthanide
177Lu presents a more favorable profile, with a shorter emission range of 2 mm as opposed to iodine-131’s 3 mm.
These radiolabeled antibodies were subsequently administered to mice harboring SK-Mel-187 and SK-Mel-16 xenograft tumors to assess their tumor uptake. In this investigation,
177Lu-CHX-A″-DTPA-vF19 and
177Lu-CHX-A″-DTPA-A33 were included as control groups. Notably, SPECT/CT imaging conducted 72 h post-injection revealed a higher specific uptake of
177Lu-ESC11 in SK-MEL-187 tumors, whereas SK-MEL-16 xenografts exhibited lower uptake compared with the control group. This study, involving a comparative analysis of the in vivo targeting attributes of human–mouse chimeric antibodies, established that in mouse models characterized by higher levels of antigen expression, the cumulative tumor uptake of the nuclide-labeled antibody could reach levels corresponding to 50% of the administered dose per gram. Conclusive in vivo experiments in mice further corroborated that the ratio of tumor-to-organ uptake pertaining to
177Lu-labeled FAP mAbs ESC11 and ESC14 surpassed that of their first-generation radionuclide-labeled FAP-targeted antibodies
[110][126]. The novel antibodies, ESC11 and ESC14, exhibited efficient internalization into FAP-expressing cells, thereby manifesting highly satisfactory in vivo targeting capabilities.
2.4.
89
Zr-Labeled F19 and B12 IgG
Clinical investigations involving first-generation FAP-targeting antibodies have provided novel insights by demonstrating the feasibility of modifying FAP-specific cancer targeting through the conjugation of toxins or chelators with FAP-specific antibodies
[134][135][153,154]. As an illustrative example, Pandya et al. prepared the radiopharmaceutical antibody conjugate [
89Zr]Zr-Df-Bz-F19 mAb for PET imaging by employing the bifunctional chelator Df-Bz-NCS to securely bind zirconium-89 (
89Zr) (
Figure 1)
[111][127].
89Zr, a radionuclide, emits β+ particles at 902 keV with an abundance of 23% and possesses a half-life of 78.4 h
[132][136][105,155]. Its attributes, characterized by high-resolution imaging, specific tissue binding, and strong signal contrast, render it a promising candidate for PET imaging applications
[137][138][156,157]. Nonetheless, certain challenges persist, primarily the susceptibility to covalent bond breakage between the chelator and the protein, which can compromise stability
[139][158]. To mitigate this concern, comprehensive in vitro characterization of [
89Zr]Zr-Df-Bz-F19 mAb was conducted. The radiolabel displayed a remarkable radioactive purity exceeding 99.5% upon synthesis completion and retained its purity at levels greater than 99.1% in human serum after 7 days of incubation at 36–37 °C. These findings affirm the robust stability of nucleoporin labeling and underscore the ability of [
89Zr]Zr-Df-Bz-F19 to maintain its structural integrity following in vivo administration.
FAP expression is documented in multiple solid cancers, yet limited knowledge exists regarding its prevalence in metastatic castration-resistant prostate cancer (mCRPC)
[140][141][142][143][159,160,161,162]. The evolving landscape of precision treatment strategies for prostate cancer was highlighted in the European Society of Medical Oncology 2022 report
[144][145][163,164]. Advancements in CRPC therapies hinge on accurate imaging modalities, lesion visualization, disease staging, and informed therapeutic decision-making
[132][105]. These findings underscore the ongoing demand for selective and sensitive imaging probes applicable to mCRPC patients.
To address the specific context of mCRPC, Hallie et al. employed a humanized antibody, initially identified by phage display, and labeled it with
89Zr. This antibody serves as a FAP-expressing tumor-selective imaging probe for PET/CT imaging in a preclinical prostate cancer xenograft model
[112][128]. The investigative process commenced with genomic and immunohistochemistry assessments to determine the expression of FAP in prostate cancer
[112][146][147][128,165,166]. Specifically, FAP localization in tissues and cells was determined via antigen–antibody binding reactions. Genomic analysis was predicated on RNA sequencing derived from primary prostate cancer patient samples or mCRPC bone and soft tissue tumor biopsies
[148][149][167,168].
Subsequent PET/CT imaging evaluations were conducted in a mouse model established through the subcutaneous injection of CWR-R1FAP cells. Notably, [
89Zr]Zr-B12 IgG demonstrated significantly enhanced tumor uptake in FAP-positive cells in mice bearing CWR-R1FAP relative to the control group injected with [
89Zr]Zr-IC IgG. This heightened tumor accumulation is attributed to improved permeability and retention efficiency, leading to a sustained presence of the [
89Zr]Zr-IC IgG probe within the tumor tissue, unlike the control probe, which exhibited rapid clearance and near-invisibility at 72 h. Compared with that in the control group, there was a heightened accumulation of [
89Zr]Zr-B12 IgG in FAP-positive tumor cells (
Figure 1)
[112][128].
In a critical extension of this work, mice were subcutaneously injected with hPrCSC-44 (an immortalized human prostate cancer stromal cell line) in combination with DU145 (a FAP-null prostate cancer cell line) to establish a xenograft mouse model. Subsequent administration of [
89Zr]Zr-B12 IgG or control [
89Zr]Zr-IC IgG in mice bearing subcutaneous hPrCSC-44/DU145 xenografts, followed by serial imaging was performed at 24, 48, 72, 96, 120, and 144 h post-injection, allowed for comprehensive PET imaging evaluation. The results showed maximum tumor uptake of [
89Zr]Zr-B12 IgG at 24 h, which surpassed control levels by 4–5 times.
2.5. Bispecific Antibodies
In the realm of tumor-targeting antibodies, an exciting development involves Hoffmann LaRoche’s bispecific antibodies RG7386 (FAP-DR5) and RO7300490 (FAP-CD40) (
Figure 1)
[114][150][151][134,171,172]. Currently in phase I clinical trials (NCT02558140, NCT04857138)
[152][153][173,174], these antibodies represent a novel approach. One of these antibodies is engineered to target FAP for precise localization, while the other is designed to interact with molecules influencing tumor apoptosis or necrosis. These bispecific antibodies work in tandem, obstructing different signaling pathways simultaneously. Compared with monoclonal antibodies, bispecific antibodies possess two specific antigen-binding sites, which endow them with stronger specificity. Consequently, they precisely target tumor cells, minimize off-target toxicity, and can orchestrate immune cell-mediated tumor eradication through dual-target signal blockade
[154][155][156][157][158][159][160][161][175,176,177,178,179,180,181,182]. This special structure confers a unique advantage to bispecific antibodies in the field of tumor therapy.
The concept of bispecific antibodies also opens up new possibilities for radionuclide labeling. While no specific studies have explored the radionuclide labeling of bispecific antibodies targeting FAP, research into combination therapies involving radionuclides and bispecific antibodies targeting other antigens has been conducted
[162][163][183,184]. For instance, Morris’ team combined radionuclides with bispecific antibodies (anti-CTLA-4 and anti-PD-L1). In a mouse model, this approach resulted in complete and enduring tumor remission, outperforming combinations involving monoclonal antibodies
[164][185]. The potential to combine or label FAP-targeted bispecific antibodies is a promising avenue that introduces a fresh dimension to radiotherapy nuclide markers for FAP-targeted tumor therapy. Such innovations hold significant potential for advancing clinical FAP-targeted tumor therapy.