Heart failure with preserved ejection fraction (HFpEF) is increasingly prevalent and now accounts for half of all heart failure cases. This rise is largely attributed to growing rates of obesity, hypertension, and diabetes. The heart, being the most energy-demanding organ, appears to have a compromised bioenergetic capacity in heart failure, affecting all phenotypes and aetiologies.
- HFpEF
- heart failure
- cardiac metabolism
- substrate utilisation
- energy production
1. Introduction
2. Normal Cardiac Metabolism
The initial steps in ATP generation are the uptake and utilisation of fuels in the myocardium. Under normal conditions, the heart sources the majority of its ATP from fatty acids (70–90%) and to a lesser extent carbohydrates (10–40%), with smaller contributions from ketone bodies and amino acids (Figure 1) [4]. The final end product of these pathways is acetyl coenzyme A which can then be fed into the tricarboxylic acid (TCA) cycle. The heart is metabolically flexible, adapting its substrate usage based on local and systemic conditions, allowing ATP generation to continue in fed, fasted, and high-demand states.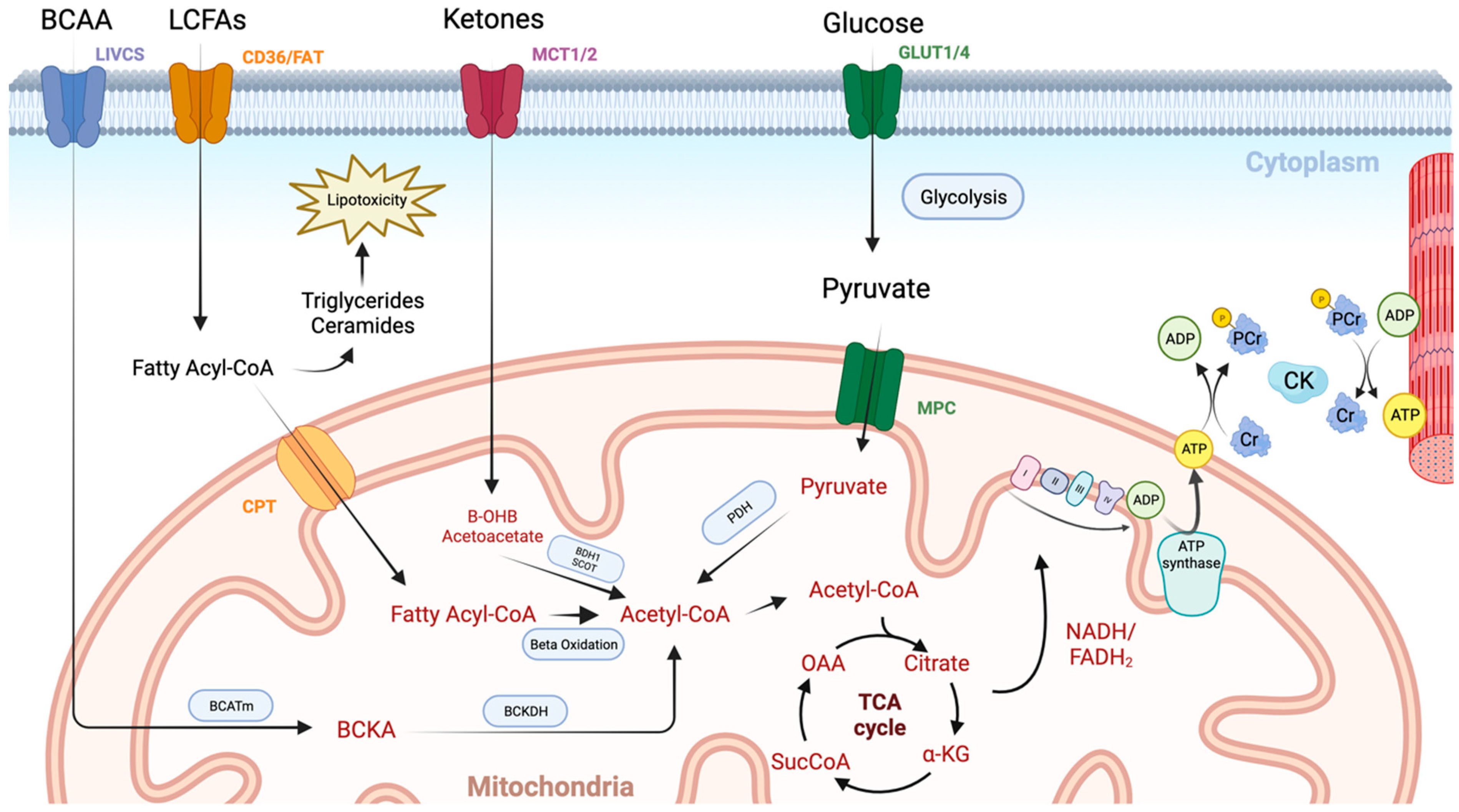
3. Cardiac Metabolism in HFpEF
3.1. Animal Studies
Much of our knowledge of myocardial metabolism in heart failure comes from ischaemia–reperfusion animal models which reliably progress to HFrEF. However, given the distinct risk factors, pathophysiological mechanisms, and clinical phenotype in HFpEF, any derangement in cardiac metabolism may be different from than observed in HFrEF. Animal models of HFpEF have, however, proven more difficult to develop, partly owing to the heterogeneity in the clinical phenotype of HFpEF patients. Early studies focused on HFpEF risk factors such as obesity, hypertension, diabetes, and ageing, with animal models of these conditions in isolation being used as surrogates for HFpEF. Whilst many of these models showed classic features of HFpEF such as diastolic dysfunction and preserved systolic function, they often lacked key clinical features such as pulmonary congestion and elevated filling pressures [8]. More advanced animal models of HFpEF have sought to combine hypertensive, metabolic, and ageing stressors and have had more success in generating a more comprehensive set of HFpEF features. For instance, a novel ‘three-hit’ HFpEF mouse model has recently been developed, with obesity induced via a high-fat diet and hypertension via an injection of desoxycorticosterone pivalate (DOCP), with ageing as the final ‘hit’. This model accurately replicates key haemodynamic and clinical features of HFpEF including diastolic dysfunction, reduced exercise tolerance, and pulmonary congestion, all whilst maintaining left ventricular ejection fraction [9]. Interestingly, hypertensive and metabolic stresses appear to have competing consequences for substrate utilisation. In single-factor hypertensive models, a shift away from fatty acid oxidation towards glucose metabolism becomes apparent [10], similar to that seen in HFrEF. Whilst traditionally this shift was believed to confer an efficiency benefit in terms of oxygen use, it is becoming more established that this increased glycolytic activity occurs to support aspartate synthesis, a key intermediate in nucleotide synthesis, which is important in driving cardiac hypertrophy [11]. In contrast, metabolic models such as db/db mice [12[12][13][14],13,14], ZDF rats [15[15][16],16], and HFD/STZ rats [17,18][17][18] have increased fatty acid oxidation, presumably due to insulin resistance and decreased access to glucose. In the previously mentioned ‘three-hit’ HFpEF model, the authors found that fatty acid utilisation was slightly increased. It may be the case that the insulin resistance present caused a reciprocal increase in fatty acid oxidation via the Randal cycle, ultimately explaining the slightly increased fatty acid oxidation. It is clear, however, that there are complex pathways governing substrate use and that the flux of these pathways is likely to change with comorbidities and therefore HFpEF phenotype. The increase in fatty acid oxidation does not appear to be sufficient to prevent accumulation of fatty acids and associated lipotoxicity. This intramyocardial lipid accumulation appears to be directly related to the development of diastolic dysfunction [19,20,21][19][20][21]. Fatty acids gain entry to the cytoplasm via the fatty acid translocase (CD36) located on the cytoplasmic membrane. Upon entry, they must cross the mitochondrial membrane in order to undergo fatty acid oxidation. Long-chain fatty acids, however, are unable to cross this membrane and require the aid of a carnitine shuttle. The key transporter in this shuttle is carnitine palmitoyltransferase 1 (CPT1), which regulates fatty acid uptake into the mitochondria and thus oxidative phosphorylation. Ketone bodies are an alternative myocardial fuel, and recent work has suggested that they contribute to myocardial metabolism in the healthy human heart [30][22]. These are synthesised from acetyl coenzyme A in hepatocytes and provide a carbon source for ATP synthesis in times of fasting, exercise, and exogenous ketone ingestion [31][23]. In HFrEF animal models, ketone body utilisation increases in what appears to be an adaptive response [32,33][24][25]. Furthermore, mice with cardio-specific knockdown of key ketolytic enzymes experienced worsening hypertrophy and systolic dysfunction [34][26], while infusion of ketone bodies appears to be protective [34,35][26][27]. Downstream of substrate utilisation, few studies have probed myocardial energetics in animal models of HFpEF [8]. Studies using 31-phosphorous magnetic resonance spectroscopy have shown decreases in PCr/ATP with obesity [36,37][28][29] and ageing [38][30] in murine models. However, data are lacking for phosphorous spectroscopy studies in two- and three-hit animal models of HFpEF. This is likely to be important in validating these newer models of HFpEF against human HFpEF. Finally, ATP generated at the mitochondria must be transferred to the myofibrils via the creatine kinase (CK) shuttle. Again, this has been studied in conditions predisposing to HFpEF rather than in the condition directly. Forward CK flux ([phosphocreatine] x CK forward rate constant) has been shown to be significantly reduced in canine and porcine models of left ventricular hypertrophy (LVH) [39,40][31][32], with greater reductions present upon development of heart failure [39][31]. In isolated perfused hearts of obese mice who exhibited diastolic dysfunction, CK flux was maintained due to elevations in CK forward rate constant [37][29]. Similarly, in a diabetic rodent model, the CK forward rate constant was increased with CK flux similar to matched controls [41][33]. Future studies should seek to investigate CK kinetics in animal models of HFpEF that combine risk factors.3.2. Human Studies
In contrast to the animal studies mentioned above, the bulk of work probing myocardial metabolism in human subjects with HFpEF has been directed at downstream ATP production. Most of this work has utilised phosphorous spectroscopy to non-invasively assess PCr/ATP ratio. Decreases in PCr/ATP in human subjects with HFrEF are well recognised [6,42[6][34][35][36],43,44], and PCr/ATP has been shown to be a better predictor of mortality than left ventricular ejection fraction [42][34]. Similarly, in HFpEF patients, a 20–27% reduction in PCr/ATP has been shown [45,46,47][37][38][39]. Moreover, PCr/ATP has been shown to be reduced in the hearts of patients with risk factors for HFpEF such as obesity [48[40][41],49], diabetes [45[37][42],50], hypertension [51][43], and ageing [52][44]. Interestingly, recent work has linked this energetic deficit in HFpEF to impaired left ventricular diastolic reserve, left atrial dilation, and pulmonary congestion during exercise, supporting the importance of energetic derangement as a key mechanism in HFpEF [45][37]. Once ATP is produced, the creatine kinase shuttle is crucial in the transportation of high-energy phosphates from mitochondria to myofibrils for utilisation. In patients with non-ischaemic cardiomyopathy, myocardial CK flux has been shown to predict heart failure outcomes independent of left ventricular ejection fraction and NYHA class [53][45]. Whilst limited data are available for CK flux in HFpEF, it has been studied in left ventricular hypertrophy (LVH) [54][46] and in obesity [48][40]. In hypertension-related LVH, the forward rate constant of myocardial CK reaction was normal, with CK flux reduced by 30% compared with healthy controls. However, in those with LVH and associated systolic dysfunction, the forward rate constant was halved and CK flux was reduced by almost two-thirds [54][46]. In obesity, the forward rate constant of the myocardial CK reaction at rest was found to be increased, yielding no overall difference in resting ATP delivery (CK flux) [48][40]. However, upon exercise, this flux was unable to be augmented, resulting in cardiopulmonary exercise intolerance [48][40]. Given exercise intolerance is a cardinal feature of HFpEF, it will be interesting to examine CK flux at rest and stress in individuals living with HFpEF. Upstream substrate utilisation has not been fully assessed in patients living with HFpEF. Studies in patients living with obesity have shown correlations between insulin resistance and increased myocardial fatty acid uptake, utilisation, and oxidation [55][47], while studies in patients with diabetic cardiomyopathy have shown increased myocardial fatty acid uptake and oxidation and decreased glucose uptake [56,57][48][49]. A mismatch between the uptake and oxidation of fatty acid species leads to their accumulation and resultant lipotoxicity. Proton (1H) spectroscopy can be used to non-invasively assess myocardial triglyceride content and has been used to show that HFpEF patients have significantly more intramyocardial fat than either HFrEF or control subjects [61][50]. Furthermore, intramyocardial fat correlated with the severity of diastolic dysfunction independently of risk factors. The extent to which myocardial lipotoxicity drives HFpEF is unknown, but it represents an attractive treatment target.4. Therapeutic Strategies
Given the metabolic derangements in HFpEF discussed above, therapeutic modulation of metabolic pathways represents an exciting new treatment strategy for HFpEF. This may involve manipulating fatty acid, glucose, or ketone oxidation (Table 1) to yield improved ‘fuel efficiency’, improving contractile function by increased ATP availability.|
Therapeutic Approach |
Rationale |
Summary |
References |
|---|---|---|---|
|
Balancing Fatty Acid Metabolism |
|||
|
Weight Loss |
Reduction in circulating lipids and myocardial steatosis. |
Not yet studied in HFpEF, but improved myocardial energetics in obesity +/− diabetes and reductions in myocardial steatosis following dietary weight loss. |
|
|
Nicotinic acid derivates |
Reduction in circulating lipids. |
No studies of niacin in HF, but acipimox (niacin derivative) has shown disappointing results in HFrEF. |
|
|
PPAR agonists (e.g., fenofibrate) |
Reduction in circulating lipids. |
No prospective studies, but post hoc analysis suggests reduced HF hospitalisations. |
|
|
SIRT6 activator (MDL-800) |
Reduction of fatty acid translocation across the endothelium. |
Protection against cardiac steatosis and diastolic function in murine model. |
[18] |
|
Angiotensin 1–7 |
Negative regulator of RAAS. |
Improved diastology and reduced myocardial steatosis with increase in cardiac lipase expression in a diabetic murine model. |
[13] |
|
Astragaloside IV |
Stimulation of fatty acid β-oxidation. |
Switch from glucose to fatty acid oxidation, with improved energetics and function in murine HFrEF model. Improved diastology in HFpEF rodent model. |
|
|
L-carnitine |
Increase fatty acid transport into the mitochondria via carnitine shuttle. |
Improvements in diastology, pulmonary congestion, and survival in hypertensive HFpEF rodent model. Reduction in symptoms and diastolic dysfunction in diastolic HF patients. |
|
|
Nicotinamide riboside |
NAD+ repletion, decreasing acetylation of key enzymes in FAO pathway. |
Improved mitochondrial function and symptoms in HFpEF murine model. |
|
|
Intralipid infusion |
Supplying myocardium with a rich fuel source which it prefers in health. |
Studied only in HFrEF patients, but improvements in energy production and LV systolic function. |
|
|
Improving Glucose Utilisation |
|||
|
Trimetazidine |
Inhibition of FAO in order to increase glucose oxidation. |
Some suggested benefit in HFrEF, ongoing DoPING-HFpEF study will evaluate in HFpEF. |
|
|
Ranolazine |
Inhibition of FAO in order to increase glucose oxidation. |
Improved haemodynamics in HFpEF patients with no change in relaxation parameters (RALI-DHF). |
|
|
Etomoxir |
Inhibiting CPT1, responsible for transport of fatty acids into the mitochondria. |
Improvement in function in 10 HFrEF patients, no studies in HFpEF, limited by neurotoxicity and hepatotoxicity. |
|
|
Perhexilline |
Inhibiting CPT1, responsible for transport of fatty acids into the mitochondria. |
Improvements in symptoms and myocardial energetics in HCM. RCT in HFpEF completed but not reported (NCT00839228). |
|
|
Ninerafaxstat |
Inhibition of FAO in order to increase glucose oxidation. |
Normalisation of myocardial energetics and improved diastolic filling and cardiac steatosis in diabetic cardiomyopathy. |
|
|
Dichloroacetate |
Inhibition of PDHK, thus increasing PDH flux and linking glycolysis with glucose oxidation. |
Preclinical HFpEF models show improvements in contractility, hypertrophy, and increased energy reserves. Concerns over neurotoxicity. |
|
|
GLP1-RA |
Increased insulin secretion and sensitivity, allowing increased glucose uptake. |
Improvements in cardiac function greater than SGLT2i in murine HFpEF model. Early results in humans hint at improvements in cardiac energetics. |
|
|
Increasing Ketone Body Utilisation |
|||
|
Ketone body supplementation |
Alternative fuel source requiring less oxygen per mole of ATP produced. |
Beneficial effects in human HFrEF and potential benefits in animal HFpEF models, clinical studies in HFpEF are awaited. |
|
|
SGLT2i |
Shown to increase circulating ketone body levels, offering alternative myocardial fuel. |
Conflicting results regarding changes in myocardial energetics in HFpEF with SGLT2i treatment. Further work is needed. |
HF: heart failure, HFpEF: heart failure with preserved ejection fraction, HFrEF: heart failure with reduced ejection fraction, PPAR: peroxisome proliferator-activated receptors, SIRT6: sirtuin 6, RAAS: renin–angiotensin–aldosterone system, FAO: fatty acid oxidation, CPT1: carnitine palmitoyltransferase 1, RCT: randomised controlled trial, PDHK: pyruvate dehydrogenase kinase, GLP1-RA: glucagon-like peptide-1 receptor agonist, SGLT2: sodium glucose transporter 2, ATP, adenosine triphosphate.
References
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc. Res. 2023, 118, 3272–3287.
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461.
- Solomon, S.D.; McMurray, J.J.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098.
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151.
- Burrage, M.K.; Lewis, A.J.; Miller, J.J.J. Functional and Metabolic Imaging in Heart Failure with Preserved Ejection Fraction: Promises, Challenges, and Clinical Utility. Cardiovasc. Drugs Ther. 2023, 37, 379–399.
- Neubauer, S.; Krahe, T.; Schindler, R.; Horn, M.; Hillenbrand, H.; Entzeroth, C.; Mader, H.; Kromer, E.P.; Riegger, G.A.; Lackner, K.; et al. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation 1992, 86, 1810–1818.
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.S.P.; Obokata, M.; Rider, O.J. Obesity and heart failure with preserved ejection fraction: New insights and pathophysiological targets. Cardiovasc. Res. 2022, 118, 3434–3450.
- Yoshii, A.; Tian, R. Remodeling of cardiac metabolism in heart failure with preserved ejection fraction. Curr. Opin. Physiol. 2022, 27, 100559.
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245.
- Gao, S.; Liu, X.P.; Li, T.T.; Chen, L.; Feng, Y.P.; Wang, Y.K.; Yin, Y.J.; Little, P.J.; Wu, X.Q.; Xu, S.W.; et al. Animal models of heart failure with preserved ejection fraction (HFpEF): From metabolic pathobiology to drug discovery. Acta Pharmacol. Sin. 2023, 45, 23–35.
- Ritterhoff, J.; Young, S.; Villet, O.; Shao, D.; Neto, F.C.; Bettcher, L.F.; Hsu, Y.W.A.; Kolwicz Jr, S.C.; Raftery, D.; Tian, R. Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis. Circ. Res. 2020, 126, 182–196.
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349.
- Mori, J.; Patel, V.B.; Abo Alrob, O.; Basu, R.; Altamimi, T.; DesAulniers, J.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Angiotensin 1–7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart Fail. 2014, 7, 327–339.
- Ko, K.Y.; Wu, Y.W.; Liu, C.W.; Cheng, M.F.; Yen, R.F.; Yang, W.S. Longitudinal evaluation of myocardial glucose metabolism and contractile function in obese type 2 diabetic db/db mice using small-animal dynamic (18)F-FDG PET and echocardiography. Oncotarget 2017, 8, 87795–87808.
- Chatham, J.C.; Seymour, A.M. Cardiac carbohydrate metabolism in Zucker diabetic fatty rats. Cardiovasc. Res. 2002, 55, 104–112.
- Wang, P.; Lloyd, S.G.; Zeng, H.; Bonen, A.; Chatham, J.C. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2102–H2110.
- Mansor, L.S.; Gonzalez, E.R.; Cole, M.A.; Tyler, D.J.; Beeson, J.H.; Clarke, K.; Carr, C.A.; Heather, L.C. Cardiac metabolism in a new rat model of type 2 diabetes using high-fat diet with low dose streptozotocin. Cardiovasc. Diabetol. 2013, 12, 136.
- Wu, X.; Liu, H.; Brooks, A.; Xu, S.; Luo, J.; Steiner, R.; Mickelsen, D.M.; Moravec, C.S.; Alexis, J.D.; Small, E.M.; et al. SIRT6 Mitigates Heart Failure with Preserved Ejection Fraction in Diabetes. Circ. Res. 2022, 131, 926–943.
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684.
- Abdurrachim, D.; Ciapaite, J.; Wessels, B.; Nabben, M.; Luiken, J.J.; Nicolay, K.; Prompers, J.J. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim. Biophys. Acta 2014, 1842, 1525–1537.
- Leggat, J.; Bidault, G.; Vidal-Puig, A. Lipotoxicity: A driver of heart failure with preserved ejection fraction? Clin. Sci. 2021, 135, 2265–2283.
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368.
- Paolisso, G.; Gambardella, A.; Galzerano, D.; D’Amore, A.; Rubino, P.; Verza, M.; Teasuro, P.; Varricchio, M.; D’Onofrio, F. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism 1994, 43, 174–179.
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616.
- Capone, F.; Sotomayor-Flores, C.; Bode, D.; Wang, R.; Rodolico, D.; Strocchi, S.; Schiattarella, G.G. Cardiac metabolism in HFpEF: From fuel to signalling. Cardiovasc. Res. 2022, 118, 3556–3575.
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079.
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684.
- Shao, D.; Kolwicz Jr, S.C.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.W.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997.
- Luptak, I.; Sverdlov, A.L.; Panagia, M.; Qin, F.; Pimentel, D.R.; Croteau, D.; Siwik, D.A.; Ingwall, J.S.; Bachschmid, M.M.; Balschi, J.A.; et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J. Mol. Cell Cardiol. 2018, 116, 106–114.
- Chiao, Y.A.; Kolwicz, S.C.; Basisty, N.; Gagnidze, A.; Zhang, J.; Gu, H.; Djukovic, D.; Beyer, R.P.; Raftery, D.; MacCoss, M.; et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging 2016, 8, 314–327.
- Ye, Y.; Gong, G.; Ochiai, K.; Liu, J.; Zhang, J. High-energy phosphate metabolism and creatine kinase in failing hearts: A new porcine model. Circulation 2001, 103, 1570–1576.
- Ye, Y.; Wang, C.; Zhang, J.; Cho, Y.K.; Gong, G.; Murakami, Y.; Bache, R.J. Myocardial creatine kinase kinetics and isoform expression in hearts with severe LV hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H376–H386.
- Bashir, A.; Coggan, A.R.; Gropler, R.J. In vivo creatine kinase reaction kinetics at rest and stress in type II diabetic rat heart. Physiol. Rep. 2015, 3, e12248.
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial Phosphocreatine-to-ATP Ratio Is a Predictor of Mortality in Patients with Dilated Cardiomyopathy. Circulation 1997, 96, 2190–2196.
- Hardy, C.J.; Weiss, R.G.; Bottomley, P.A.; Gerstenblith, G. Altered myocardial high-energy phosphate metabolites in patients with dilated cardiomyopathy. Am. Heart J. 1991, 122 Pt 1, 795–801.
- Ingwall, J.S.; Weiss, R.G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004, 95, 135–145.
- Burrage, M.K.; Hundertmark, M.; Valkovič, L.; Watson, W.D.; Rayner, J.; Sabharwal, N.; Ferreira, V.M.; Neubauer, S.; Miller, J.J.; Rider, O.J.; et al. Energetic Basis for Exercise-Induced Pulmonary Congestion in Heart Failure with Preserved Ejection Fraction. Circulation 2021, 144, 1664–1678.
- Mahmod, M.; Pal, N.; Rayner, J.; Holloway, C.; Raman, B.; Dass, S.; Levelt, E.; Ariga, R.; Ferreira, V.; Banerjee, R.; et al. The interplay between metabolic alterations, diastolic strain rate and exercise capacity in mild heart failure with preserved ejection fraction: A cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2018, 20, 88.
- Phan, T.T.; Abozguia, K.; Nallur Shivu, G.; Mahadevan, G.; Ahmed, I.; Williams, L.; Dwivedi, G.; Patel, K.; Steendijk, P.; Ashrafian, H.; et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J. Am. Coll. Cardiol. 2009, 54, 402–409.
- Rayner, J.J.; Peterzan, M.A.; Watson, W.D.; Clarke, W.T.; Neubauer, S.; Rodgers, C.T.; Rider, O.J. Myocardial Energetics in Obesity: Enhanced ATP Delivery Through Creatine Kinase with Blunted Stress Resonse. Circulation 2020, 141, 1152–1163.
- Rider, O.J.; Francis, J.M.; Ali, M.K.; Holloway, C.; Pegg, T.; Robson, M.D.; Tyler, D.; Byrne, J.; Clarke, K.; Neubauer, S. Effects of catecholamine stress on diastolic function and myocardial energetics in obesity. Circulation 2012, 125, 1511–1519.
- Levelt, E.; Mahmod, M.; Piechnik, S.K.; Ariga, R.; Francis, J.M.; Rodgers, C.T.; Clarke, W.T.; Sabharwal, N.; Schneider, J.E.; Karamitsos, T.D.; et al. Relationship between Left Ventricular Structural and Metabolic Remodeling in Type 2 Diabetes. Diabetes 2016, 65, 44–52.
- Lamb, H.J.; Beyerbacht, H.P.; van der Laarse, A.; Stoel, B.C.; Doornbos, J.; van der Wall, E.E.; de Roos, A. Diastolic dysfunction in hypertensive heart disease is associated with altered myocardial metabolism. Circulation 1999, 99, 2261–2267.
- Hollingsworth, K.G.; Blamire, A.M.; Keavney, B.D.; Macgowan, G.A. Left ventricular torsion, energetics, and diastolic function in normal human aging. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H885–H892.
- Bottomley, P.A.; Panjrath, G.S.; Lai, S.; Hirsch, G.A.; Wu, K.; Najjar, S.S.; Steinberg, A.; Gerstenblith, G.; Weiss, R.G. Metabolic rates of ATP transfer through creatine kinase (CK Flux) predict clinical heart failure events and death. Sci. Transl. Med. 2013, 5, 215re3.
- Smith, C.S.; Bottomley, P.A.; Schulman, S.P.; Gerstenblith, G.; Weiss, R.G. Altered Creatine Kinase Adenosine Triphosphate Kinetics in Failing Hypertrophied Human Myocardium. Circulation 2006, 114, 1151–1158.
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.; Marsala, J.; Meyer, T.; et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004, 109, 2191–2196.
- Rijzewijk, L.J.; van der Meer, R.W.; Lamb, H.J.; de Jong, H.W.; Lubberink, M.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Twisk, J.W.; Heine, R.J.; et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: Studies with cardiac positron emission tomography and magnetic resonance imaging. J. Am. Coll. Cardiol. 2009, 54, 1524–1532.
- Herrero, P.; Peterson, L.R.; McGill, J.B.; Matthew, S.; Lesniak, D.; Dence, C.; Gropler, R.J. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J. Am. Coll. Cardiol. 2006, 47, 598–604.
- Wu, C.K.; Lee, J.K.; Hsu, J.C.; Su, M.Y.M.; Wu, Y.F.; Lin, T.T.; Lan, C.W.; Hwang, J.J.; Lin, L.Y. Myocardial adipose deposition and the development of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2020, 22, 445–454.
- Rider, O.J.; Francis, J.M.; Tyler, D.; Byrne, J.; Clarke, K.; Neubauer, S. Effects of weight loss on myocardial energetics and diastolic function in obesity. Int. J. Cardiovasc. Imaging 2013, 29, 1043–1050.
- Hammer, S.; Snel, M.; Lamb, H.J.; Jazet, I.M.; van der Meer, R.W.; Pijl, H.; Meinders, E.A.; Romijn, J.A.; de Roos, A.; Smit, J.W. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J. Am. Coll. Cardiol. 2008, 52, 1006–1012.
- Halbirk, M.; Nørrelund, H.; Møller, N.; Schmitz, O.; Gøtzsche, L.; Nielsen, R.; Nielsen-Kudsk, J.E.; Nielsen, S.S.; Nielsen, T.T.; Eiskjær, H.; et al. Suppression of circulating free fatty acids with acipimox in chronic heart failure patients changes whole body metabolism but does not affect cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1220–H1225.
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Hesse, B.; Airaksinen, K.J.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137.
- Ferreira, J.P.; Vasques-Nóvoa, F.; Ferrão, D.; Saraiva, F.; Falcão-Pires, I.; Neves, J.S.; Sharma, A.; Rossignol, P.; Zannad, F.; Leite-Moreira, A. Fenofibrate and Heart Failure Outcomes in Patients with Type 2 Diabetes: Analysis From ACCORD. Diabetes Care 2022, 45, 1584–1591.
- Dong, Z.; Zhao, P.; Xu, M.; Zhang, C.; Guo, W.; Chen, H.; Tian, J.; Wei, H.; Lu, R.; Cao, T. Astragaloside IV alleviates heart failure via activating PPARα to switch glycolysis to fatty acid β-oxidation. Sci. Rep. 2017, 7, 2691.
- Lin, X.; Wang, Q.; Sun, S.; Xu, G.; Wu, Q.; Qi, M.; Bai, F.; Yu, J. Astragaloside IV promotes the eNOS/NO/cGMP pathway and improves left ventricular diastolic function in rats with metabolic syndrome. J. Int. Med. Res. 2020, 48, 0300060519826848.
- Omori, Y.; Ohtani, T.; Sakata, Y.; Mano, T.; Takeda, Y.; Tamaki, S.; Tsukamoto, Y.; Kamimura, D.; Aizawa, Y.; Miwa, T.; et al. L-Carnitine prevents the development of ventricular fibrosis and heart failure with preserved ejection fraction in hypertensive heart disease. J. Hypertens. 2012, 30, 1834–1844.
- Serati, A.R.; Motamedi, M.R.; Emami, S.; Varedi, P.; Movahed, M.R. L-carnitine treatment in patients with mild diastolic heart failure is associated with improvement in diastolic function and symptoms. Cardiology 2010, 116, 178–182.
- Potnuri, A.G.; Purushothaman, S.; Saheera, S.; Nair, R.R. Mito-targeted antioxidant prevents cardiovascular remodelling in spontaneously hypertensive rat by modulation of energy metabolism. Clin. Exp. Pharmacol. Physiol. 2022, 49, 35–45.
- Watson, W.D.; Green, P.G.; Lewis, A.J.; Arvidsson, P.; De Maria, G.L.; Arheden, H.; Heiberg, E.; Clarke, W.T.; Rodgers, C.T.; Valkovič, L.; et al. Retained Metabolic Flexibility of the Failing Human Heart. Circulation 2023, 148, 109–123.
- Fragasso, G.; Palloshi, A.; Puccetti, P.; Silipigni, C.; Rossodivita, A.; Pala, M.; Calori, G.; Alfieri, O.; Margonato, A. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J. Am. Coll. Cardiol. 2006, 48, 992–998.
- Fragasso, G.; Salerno, A.; Lattuada, G.; Cuko, A.; Calori, G.; Scollo, A.; Ragogna, F.; Arioli, F.; Bassanelli, G.; Spoladore, R.; et al. Effect of partial inhibition of fatty acid oxidation by trimetazidine on whole body energy metabolism in patients with chronic heart failure. Heart 2011, 97, 1495–1500.
- van de Bovenkamp, A.A.; Bakermans, A.J.; Allaart, C.P.; Nederveen, A.J.; Kok, W.E.M.; Van Rossum, A.C.; Handoko, M.L. TrimetaziDine as a Performance-enhancING drug in heart failure with preserved ejection fraction (DoPING-HFpEF): Rationale and design of a placebo-controlled cross-over intervention study. Neth. Heart J. 2020, 28, 312–319.
- Maier, L.S.; Layug, B.; Karwatowska-Prokopczuk, E.; Belardinelli, L.; Lee, S.; Sander, J.; Lang, C.; Wachter, R.; Edelmann, F.; Hasenfuss, G.; et al. RAnoLazIne for the treatment of diastolic heart failure in patients with preserved ejection fraction: The RALI-DHF proof-of-concept study. JACC Heart Fail. 2013, 1, 115–122.
- Schmidt-Schweda, S.; Holubarsch, C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 2000, 99, 27–35.
- Beadle, R.M.; Williams, L.K.; Kuehl, M.; Bowater, S.; Abozguia, K.; Leyva, F.; Yousef, Z.; Wagenmakers, A.J.; Thies, F.; Horowitz, J.; et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015, 3, 202–211.
- Hundertmark, M.; Siu, A.G.; Matthews, V.; Lewis, A.J.; Grist, J.T.; Patel, J.; Chamberlin, P.; Sarwar, R.; Yavari, A.; Frenneaux, M.P.; et al. A phase 2a trial investigating ninerafaxstat—A novel cardiac mitotrope for the treatment of diabetic cardiomyopathy (IMPROVE-DiCE). Eur. Heart J. 2022, 43 (Suppl. S2), ehac544-246.
- Bøgh, N.; Hansen, E.S.; Omann, C.; Lindhardt, J.; Nielsen, P.M.; Stephenson, R.S.; Laustsen, C.; Hjortdal, V.E.; Agger, P. Increasing carbohydrate oxidation improves contractile reserves and prevents hypertrophy in porcine right heart failure. Sci. Rep. 2020, 10, 8158.
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430.
- Withaar, C.; Meems, L.M.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124.
- Chowdhary, A.; Thirunavukarasu, S.; Jex, N.; Akkaya, S.; Kotha, S.; Giannoudi, M.; Procter, H.; Kellman, P.; Greenwood, J.; Plein, S.; et al. Abstract 12365: Liraglutide Improves Myocardial Stress Perfusion and Energetics in Patients with Type 2 Diabetes—A Randomised, Single Centre, Open Label, Cross-Over Drug Trial. Circulation 2023, 148 (Suppl. S1), A12365.
- Liao, S.; Tang, Y.; Yue, X.; Gao, R.; Yao, W.; Zhou, Y.; Zhang, H. β-Hydroxybutyrate Mitigated Heart Failure with Preserved Ejection Fraction by Increasing Treg Cells via Nox2/GSK-3β. J. Inflamm. Res. 2021, 14, 4697–4706.
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment with the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141.
- Gaborit, B.; Ancel, P.; Abdullah, A.E.; Maurice, F.; Abdesselam, I.; Calen, A.; Soghomonian, A.; Houssays, M.; Varlet, I.; Eisinger, M.; et al. Effect of empagliflozin on ectopic fat stores and myocardial energetics in type 2 diabetes: The EMPACEF study. Cardiovasc. Diabetol. 2021, 20, 57.
- Thirunavukarasu, S.; Jex, N.; Chowdhary, A.; Hassan, I.U.; Straw, S.; Craven, T.P.; Gorecka, M.; Broadbent, D.; Swoboda, P.; Witte, K.K.; et al. Empagliflozin Treatment Is Associated with Improvements in Cardiac Energetics and Function and Reductions in Myocardial Cellular Volume in Patients with Type 2 Diabetes. Diabetes 2021, 70, 2810–2822.
- Hundertmark, M.J.; Adler, A.; Antoniades, C.; Coleman, R.; Griffin, J.L.; Holman, R.R.; Lamlum, H.; Lee, J.; Massey, D.; Miller, J.J.; et al. Assessment of Cardiac Energy Metabolism, Function, and Physiology in Patients with Heart Failure Taking Empagliflozin: The Randomized, Controlled EMPA-VISION Trial. Circulation 2023, 147, 1654–1669.