Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Raffaele Serra and Version 2 by Peter Tang.
The new respiratory infectious disease coronavirus disease 2019 (COVID-19) that originated in Wuhan, China, in December 2019 and caused by a new strain of zoonotic coronavirus, named severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2). Most of the deceased patients had pre-existing comorbidities; over 20% had chronic kidney disease (CKD). Furthermore, although SARS-CoV-2 infection is characterized mainly by diffuse alveolar damage and acute respiratory failure, acute kidney injury (AKI) has developed in a high percentage of cases.
- SARS-CoV-2
- acute kidney injury
- dialysis
- renal transplantation
- pandemic
1. Introduction
By December 2019, unusual cases of pneumonia had been reported in the city of Wuhan, located in central China’s Hubei province. On January 12th 2020, the World Health Organization (WHO) stated that the disease was caused by a novel coronavirus, named severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2), belonging to the β-coronavirus cluster, which also includes the severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) viruses [1][2][1,2].
The resulting SARS-CoV-2 related disease was defined as the novel coronavirus disease 2019, or COVID-19, that rapidly spread throughout China, followed by an increasing number of cases in all continents with the exception of Antarctica, resulting in a global pandemic.
The data of the global burden of COVID-19 are impressive. Indeed, currently (available data up to 23 July 2020) the coronavirus is affecting 212 countries and territories around the world, with over 15 million cases and over 630,000 confirmed deaths (global mortality: 5.4%) [3][4][3,4].
The Americas and Europe are the most affected continents in the world. In the United States of America (U.S.A.), 4,103,674 cases and over 146,000 deaths have been reported. In Europe, many outbreaks are steadily rising in Russia, Spain, UK and Italy, with over 200,000 overall deaths in the continent. However, in China the rate of new cases has progressively decreased substantially since March [3]. The pandemic has also had a severe economic impact. In fact, to help improve the clinical management of patients infected with the virus, Europe has mobilized more than 10 million euros from research funding and the United Kingdom has also invested £20,000,000 to allow for the development of a COVID-19 vaccine [5]. It has also been estimated that in the U.S.A., the health care costs for a single infected patient is over $3000, resulting in an overall predicted expenditure of $654.0 billion over the entire period of the pandemic [6][7][6,7].A flu-like syndrome of mild severity has been observed in most cases (80%) of COVID-19 [8][9][8,9], but in 20% of cases there have been other complications such as interstitial pneumonia with a variable degree of respiratory failure [10] as well as thromboembolic complications, including venous thromboembolism (VTE), ischemic stroke and acute coronary syndrome (ACS)/myocardial infarction [11]. Although the principal features associated with COVID-19 are diffuse alveolar damage and acute respiratory failure, kidney impairment has also often developed, with the frequent onset of acute kidney injury (AKI) in patients infected by SARS-CoV-2. In addition, more than 20% of deceased patients were affected by chronic kidney disease (CKD) [11].
2. Epidemiology
Globally, over 15 million confirmed cases of COVID-19 have been reported to date (23 July 2020) [4]. Such a large number of affected patients have been caused by the way in which the virus is transmitted. COVID-19 is more contagious when compared to MERS and SARS since the virus spreads by human-to-human transmission via direct or fecal contact or droplets [7]. Furthermore, the possibility that the virus may be transmitted by asymptomatic individuals or by individuals within the incubation period, may also explain the high contagiousness of the disease [12]. COVID-19 infection is thought to have an incubation period up to 14 days following exposure, with most symptoms showing around four to five days after exposure [13]. However, case reports with longer incubation periods (up to 27 days) have also been reported [11]. An updated publication confirmed these findings, with the incubation lasting as long as 24 days (range: 0–24 days; median: 3.0 days) [7]. Nonetheless, the onset of symptoms is variable amongst infected patients, and the interval during which an individual with COVID-19 is contagious remains uncertain. It appears that SARS-CoV-2 can be transmitted prior to the development of symptoms and throughout the course of illness [14]. The infection can involve all age groups, including children, and, moreover, males suffer a disproportionately higher number of deaths than females according to data from cohorts of patients in China, Italy and the United States [15][16][15,16]. Patients admitted at the Tongji Hospital, the major endemic area, and positive for COVID-19, showed a median age of over 60-years and a large part (up to 42%) were affected by 1 or more comorbidities, including diabetes mellitus, cardiovascular disease, hypertension, chronic lung disease, cancer, chronic kidney disease, immunocompromising conditions, severe obesity (body mass index ≥ 40) and liver disease [17][18][17,18]. Similar evidence was derived from a cohort of 5700 hospitalized patients on the other side of the world, namely in the New York city area in the U.S.A. [15]. Moreover, the data of 333 children with confirmed SARS-CoV-2 from 11 case series were analyzed [19]. Intriguingly, despite the recent evidence that children have the same risk for infection than adults, few cases have been admitted to intensive care units (3%) and only a few deaths have been registered. The less-severe infection has been principally related to the fact that children have a stronger innate immune response and a minor prevalence of comorbidities (arterial hypertension, cardiovascular diseases) than adults [20]. Most patients also showed an inflammatory status and coagulopathies with elevated levels of high-sensitivity C-reactive protein and serum lactose dehydrogenase [8]. Interestingly, up to 15% of the hospitalized COVID-19 patients had at least one kidney abnormality represented by increased blood urea nitrogen or reduced estimated glomerular filtration rate (eGFR) which is the best marker of kidney function. Moreover, findings from different cohorts of hospitalized patients showed that 26–63% of patients presented proteinuria at admission or developed proteinuria during their stay in hospital, proteinuria being considered the most recognized sign of kidney damage [7][8][7,8]. An individual risk profile also found that COVID-19 patients with kidney abnormalities, compared with those with normal renal function at admission, were more likely to be males, with advanced age and with a worse coagulation profile [8]. In addition, a meta-analysis [21] has confirmed that CKD is associated with an enhanced risk of COVID-19 infection. As previously mentioned, an urgent epidemiological effort has been undertaken to understand what risk factors are responsible for the principal outcome associated with COVID-19, i.e., mortality. From Chinese studies, mortality rates were increasing along age categories (mortality rate of 1.3% in the 50–59 range, 3.6% in the 60–69 range, 8% in the 70–79 range and 14.8% in the ≥range 80 years), the presence of cardiovascular diseases (10.5%), diabetes (7.3%), chronic respiratory diseases (6.3%), arterial hypertension (6%) and neoplasms (mortality 5.6%) [22][23][22,23]. In Italy, data from ISS—i.e., the Italian Health Institute—indicates that 1% of the patients who died did not suffer from any other diseases, 26% had only one disease, 26% had 2 diseases and 47% had 3 or more pathologic conditions. The most common chronic preexisting diseases in deceased patients were: arterial hypertension (70%), followed by diabetes mellitus (31.7%), chronic kidney disease CKD (23.1%), atrial fibrillation (22.5%), chronic obstructive pulmonary disease (COPD) (18.1%), the presence of an active cancer within the previous 5 years (16.8%), ischemic heart disease (16%) and obesity 10% [20]. It is very remarkable that CKD was present in more than 20% of the deceased patients due to COVID-19, also surpassing, in prevalence, those affected by COPD and/or those with an active cancer within the last 5 years. In addition to the presence of CKD, it also has been demonstrated that COVID-19 patients are at increased risk of developing acute kidney injury (AKI), a clinical condition that is associated with unfavorable outcomes, including mortality [8][24][8,24]. The incidence of AKI in COVID-19 patients is similar to that found in SARS patients. This sudden loss of kidney function is strongly associated with increased mortality and morbidity and is a complication that can occur during the progression of COVID-19 in patients suffering from kidney disease as well as in those who are not [8][23][25][8,23,25]. From the analysis of several studies, this incidence varies from 0.5% to 23%, with an interval from baseline visit to the onset of AKI of 7–15 days in median [25][26][25,26]. Patients who developed AKI had a more critical prognosis, in terms of mortality rate compared with those who only had chronic illness as comorbidity (AKI increased the risk of death by 5.3 times in these patients) [27]. All these observations suggest that AKI could be one of the risk factors for mortality in COVID-19 patients, and a recent meta-analysis of 26 studies suggested that mortality from AKI may, in fact, be 13 times higher [28]. Overall, either the presence of CKD at hospital admission or the development of AKI during the COVID-19 infection have been both recognized as two independent risk factors of mortality [8][29][30][8,29,30]. COVID-19 has also affected patients on renal replacement treatments (RRT). The Italian Society of Nephrology (S.I.N.) report found 521 patients (2.8%) positive for SARS-CoV-2 in a population of 17,848 patients undergoing hemodialysis, 18 patients (0.65%) were positive in a population of 2252 patients treated with peritoneal dialysis. In positive RRT patients, 54% were dialyzed at their own dialysis center, 18% required hospitalization in sub-intensive care unit and 4.7% were dialyzed in an intensive care unit. Death occurred in a non-trivial proportion of cases (25.8%) [31]. Indeed, data from the Dialysis Units of Piedmont and Aosta Valley (two Italian Northern West regions) found that absolute mortality risk was higher in males than in females (31.1% vs. 4%, respectively) and in presence of cardiovascular disease (29.9% vs. 10.7%) whereas it was not related to dialysis vintage. Diabetes was a risk factor in transplanted patients (66.7% vs. 13.6%). A history of neoplasia was also associated with an increase in risk of death (40% vs. 19%) [32]. These data demonstrate how hemodialysis patients are particularly susceptible to COVID-19 infection.3. Etiology
Coronaviruses are a group of single-stranded RNA viruses (ssRNAs) with positive polarity, belonging to the Coronaviridae family [33]. Until 2019, six coronavirus strains, which were able to infect humans, were known. Four strains usually circulate in the human population causing mild respiratory infections [34]. In 2003 and 2012, the first two zoonotic strains of coronaviruses capable of infecting humans through an animal were identified. These have caused severe lung syndromes in recent times: SARS in 2003 and MERS in 2012 [34]. Studies carried out on these strains showed that zoonotic coronaviruses use the bat as the primary host and are transmitted to humans through an intermediate host, represented by the civet for SARS and the camel for MERS [35]. However, some bat coronaviruses have been recently identified as being able to infect human cells without the need for an intermediate host [35]. The new respiratory infection originating from Wuhan (Hubei Province) in China that spread rapidly to the rest of the country, was initially traced back to Huanan Seafood Wholesale Market [13]; but only in January 2020 was a strain of coronavirus, which was unknown, identified as the responsible pathogen of the infection. It was named as SARS-CoV-2 and belongs to the β-coronavirus cluster that includes SARS-CoV and MERS-CoV [2]. SARS-CoV-2 is the third strain of zoonotic coronaviruses currently known [1]. Subsequently, it was documented that SARS-CoV-2 is a chimaeric virus resulting from pre-existing viruses: a bat coronavirus and another coronavirus of unknown origin [36]. Its genomic sequence corresponds to the bat coronavirus with an 88% identity [37] and the pangolin coronavirus with a 99% identity [38], although the genetic analysis performed on the pangolin coronavirus did not involve the whole viral genome, but a specific site known as the receptor binding domain [37]. However, it emerged that SARS-CoV-2 and the pangolin coronavirus did not share the same structural characteristics [39]. Therefore, pangolin was identified as the intermediate species of transition from bat to humans rather than directly responsible for the SARS-CoV-2 pandemic [3][11][3,11].4. Pathophysiology
To understand the pathophysiological mechanism of SARS-CoV-2, its genomic sequence was compared to the other two similar coronaviruses, SARS-CoV and MERS-CoV. In fact, it emerged that SARS-CoV-2 has a sequence identity of 79% with SARS-CoV and 50% with MERS-CoV [37][40][37,40]. Upon analysis of certain proteins, such as the coronavirus main proteinase (3CLpro), papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp), it was observed that the sequence identity value between SARS-CoV and SARS-CoV-2 is 96% [41]. Therefore, an analogy between the physiopathological mechanisms of SARS-CoV and SARS-CoV-2 has been hypothesized [42]. Many studies reported that SARS-CoV-2, like SARS-CoV, uses Angiotensin converting enzyme 2 (ACE-2) to enter target cells [43][44][45][43,44,45]. ACE-2 is a carboxypeptidase expressed on the cell surface which cleaves Angiotensin I (Ang I) into Angiotensin 1–9 and Angiotensin II (Ang II) into Angiotensin 1–7, counteracting the vasoconstrictor, proliferative and fibrotic effects of Angiotensin II generated by Angiotensin converting enzyme (ACE) [46]. Single-cell RNA sequencing analysis demonstrated a wide distribution of ACE-2 in different tissues [12][47][12,47] and histochemical staining then confirmed these data [48]. However, since low levels of ACE-2 expression were found on several cells types [47], it was supposed that cellular interaction and internalization by SARS-CoV-2did not depend only on ACE-2, but depended also on other auxiliary cell membrane receptors and proteins. In fact, it is recognized that ssRNA viruses tend to have multiple receptors [49]. Qi et al. analyzed the expression of ACE-2 on 119 cell types from 13 human tissues and the coexpression characteristics of the ssRNA human viral receptors and membrane proteins. Pearson correlation analysis of gene expression matrices showed 94 genes were found to be significantly correlated with ACE-2. Among these, the coding genes of the peptidases alanylaminopeptidase (ANPEP), glutamyl aminopeptidase (ENPEP) and dipeptidyl peptidase 4 (DPP 4) showed the highest correlation with ACE-2 [12]. While both ANPEP and DPP4 are already known as a target receptors for other coronaviruses (human coronavirus 229E, swine epidemic diarrhea virus, canine coronavirus, feline coronavirus, for ANPEP and MERS-CoV for DPP4), the relationship between ENPEP and viral infection is not yet known [12]. ENPEP is a member of the M1 family of endopeptidases which are mammalian type II integral membrane zinc-containing endopeptidases. It is mainly expressed in the terminal ileum and in the renal cortex and plays a role in the catabolic pathway of the Renin-Angiotensin system (RAAS), in the regulation of blood pressure and in the formation of blood vessels [50]. While the observations of Qi et al. suggest ENPEP may be a coronavirus receptor, further investigation is needed to confirm this [12]. However, the low expression of ACE-2 on the cell surface could also be interpreted as a viral defense mechanism. In the past, the down regulation of ACE-2 had been correlated with faster cell-cell spread of human coronaviruses [51] and with more severe clinical manifestations [52][53][52,53]. Guzzi et al., in a recent study, hypothesized that even for COVID-19 the down regulation of ACE-2 could be a mechanism induced by SARS-CoV-2 to obtain a faster intercellular diffusion [54]. Studies carried out to understand the effect of angiotensin receptor blocker (ARB) drugs in patients with COVID-19, have suggested mechanisms through which the upregulation of ACE-2 may be protective during SARS-CoV-2 infection [55]. ARBs, in fact, greatly increase the cellular expression of ACE-2 [56]. However, since SARS-CoV-2-ACE-2 interaction represents the first step of a chain of events, if the upregulation of ACE-2 is not followed by the increase of certain cell proteases essential for internalization and viral activation, it would only result in the sequestration of SARS-CoV-2 on the cell membrane limiting viral infection [35]. Furthermore, the metalloproteinase ADAM17 can act upon the membrane-bound ACE-2, leading to the release of a soluble form of ACE-2. If the increased expression of ACE-2 correlates with an increase in soluble ACE-2, this might act as a decoy receptor for SARS-CoV-2 by limiting viral entry into target cells [35]. Upon analysis of the SARS-CoV-2-ACE-2 interaction, there was confirmation that this occurs through the spike glycoprotein expressed on the viral envelope, being the same for all coronaviruses [35]. The coronavirus spike protein is composed of an intracellular segment, a transmembrane segment and a large ectodomain formed by an S1 subunit for interaction with the target receptor and an S2 subunit for fusion between the viral and cell membrane [57]. Subunit S1 consists of four domains, one N terminal domain (NTD) and three C-terminal domains (CTD1, CTD2 and CTD3). The cell receptor and the viral protein bind through the receptor-binding domain (RBD), located in the CTD1 domain in the case of SARS-CoV [58]. Experiments undertaken to investigate virus-receptor interaction with resolution at the atomic level showed that SARS-CoV and SARS-CoV-2 had a high sequence similarity (89.2%) and sequence identity (73.7%) [14][22][14,22]. However, a more targeted evaluation of SARS-CoV-2 RBD revealed peculiar characteristics that are probably responsible for the greater diffusion compared with SARS-CoV. Wan et al. found that the SARS-CoV-2 RBD has a single mutation that improves its binding affinity with ACE-2 [59]. Heet al. have shown that the characteristics of SARS-CoV-2 RBD make the virus more soluble, therefore capable of binding ACE-2 more easily, but also more stable, therefore able to survive at high temperatures [42]. At the same time, the SARS-CoV-2 RBD has greater flexibility than the SARS-CoV RBD, especially near the binding site. For this reason, SARS-CoV-2 is much more sensitive to temperature than SARS-CoV in terms of the RBD-ACE-2 bond and this would cause the decrease in infectivity with increasing temperatures [42]. The SARS-CoV RBD-ACE-2 binding induces conformational changes in the S2 subunit, such as to induce the fusion between the viral membrane and the cell. A low pH and pH-dependent endosomal cysteine proteases called cathepsins facilitate endosomal cell entry of the virus. Furthermore, the S protein is cleaved into the S1 and S2 subunits by the host transmembrane cell proteases [60], which are necessary for the entry of the virus through the cell surface non-endosomal pathway [61]. Hoffmann et al. have shown that in particular SARS-CoV-2 S protein depends on the cellular protease Transmembrane Serine Protease 2 (TMPRSS2) for priming [62]. Therefore, the coexpression of ACE-2 and TMPRSS2 is a determining factor for the entry of SARS-CoV-2 into the host cells [63]. After entering the cell and becoming activated, SARS-CoV-2 uses the endogenous transcription mechanism of the cells to replicate and spread [60]. Cells infected by SARS-CoV-2 can recruit and modulate immune cells through the secretion of chemokines or other cytokines [12]. The role of macrophages remains to be defined. In fact, the interaction between macrophages and cells expressing ACE-2 is known, suggesting a primary role of macrophages as a sentinel during viral infection [12]. A recent study, however, has shown a down regulation of mitochondrial proteins that interact with SARS-CoV-2. This mechanism could be interpreted as a process through which the virus prevents apoptosis induced by mitochondria [54].5. Pathophysiology of Kidney Damage Induced by SARS-CoV-2
The expression of ACE-2 has been shown not only in the lung but also in the liver, stomach, ileum, colon, esophagus and kidney [47]. These data associated with the evidence that AKI (7%), myocardial dysfunction with acute cardiovascular events (12%) and gastrointestinal disorders are among the most frequent clinical manifestations of COVID-19 [23], suggesting that SARS-CoV-2 can infect these organs. However, whether SARS-CoV-2 replication occurs in these organs causing functional damage and contributing to the systemic spread of the virus is not yet clear. Zou et al. [47], stratifying the human organs in high and low risk according to the level of ACE-2 expression, have shown that the kidney is very vulnerable to SARS-CoV-2infection. Qi et al. [12], through single cell RNA sequencing studies, have shown that ACE-2 in the kidney is expressed primarily by renal proximal tubular cells (~82%) and also to a lesser extent by cells of the intercalated duct, main cells of the collecting duct, renal distal tubular cells, glomerular parietal epithelium cells and immune cells (8%) [12]. Pan et al. [63], using single cell transcriptome analysis, confirmed the cellular co-expression of ACE-2and TMPRRS genes in proximal tubule cells and podocytes. Batlle et al. have also suggested whether the expression of other cellular TMPRSSs other than TMPRSS2 (such as TMPRSS 4, 5 or 9) may also play a role in the priming step [64]. Studies showed that the co-expression of the ACE-2 receptor and TMPRSS genes in kidney epithelial cells was as significant as in the lung, esophagus, small intestine and colon, suggesting that the kidney might also be an important target organ for SARS-CoV-2 [63]. In particular, a high co-expression of ACE-2 and TMPRSS genes was found in podocytes and proximal rectangular tubular cells [63]. Other noteworthy observations include the presence ofSARS-CoV-2 nucleocapsid protein in renal tubular structures and virus-like particles in podocytes and renal tubular epithelial cells, as observed by electron microscopy [65][66][65,66]. Together these observations suggest the virus may cause AKI through a direct cytopathic effect on kidney cells. In particular, it is conceivable that the virus may enter the kidney by invading the podocytes first, thereby gaining access to the tubular fluid and thence to the proximal tubule cells where it may bind to ACE-2 [64]. The viral replication in podocytes and the resulting damage could explain the proteinuria and hematuria reported in a high percentage of COVID-19 patients [8][27][8,27]. However, if the renal dysfunction is caused only by direct damage of SARS-CoV-2 or is secondary also to other systemic processes triggered by the virus, it has not been well defined yet. Diao et al. [65] observed that kidney damage associated with COVID-19 is an acute tubular necrosis induced directly by SARS-CoV-2 during infection and replication, but also indirectly through the complex immune mechanisms triggered by cellular damage. In fact, the histopathological examination performed on kidney specimens, obtained from autopsy of COVID-19 patients with renal function impairment, showed viral antigens in the cytoplasm of the tubular cells, but also a strong presence of CD68+ macrophages in the tubulo-interstitium and strong C5b-9 depositions on the apical brush border of tubular epithelial cells (TECs). This suggested that proinflammatory cytokines derived from macrophages in the tubulo-interstitium and complement-mediated mechanisms resulting from cell damage participate in the pathogenesis of tubulo-interstitial damage. In fact, despite the infiltration of infected tissue by host immune cells in order to contain viral replication, the hyperactivation of these immune cells may lead to fibrosis, epithelial cell apoptosis and cause microvasculature damage [67][68][69][67,68,69]. Studies performed on SARS-CoV suggested that AKI in SARS patients was the result of specific pathogenic conditions, such as the cytokine release syndrome [70], rather than active viral replication in the kidney. In consideration of the analogy between SARS-CoV and SARS-CoV-2, flow cytometry was used to study the immune phenotype and the function of peripheral blood mononuclear cells in COVID-19 patients [71]. Studies showed that patients infected by SARS-CoV-2 showed lymphopenia, mainly related to the significant reduction in absolute T cell counts, particularly cytotoxic T lymphocytes (CD8+), increased neutrophil counts and elevated levels of proinflammatory cytokines. In particular, high levels of interleukin (IL)-2, IL-6, IL-10 and interferon (IFN)-γ were observed. Therefore, it has been speculated that a loss of T cells during the viral infection may result in enhanced inflammatory responses [72]. In fact, it is known that T cells are important for dampening overactive innate immune responses during viral infection [73]. In accordance with this hypothesis, it has been observed that when the T cell count drops the serum levels of IL-2, IL-4, IL-10, tumour necrosis factor (TNF)-α and interferon (IFN)-γ reach their peaks [72]. Another important finding is that COVID-19 patients with more severe clinical manifestations have higher serum concentrations of IL-6 and lower IFN-γ than mild forms. This is mainly due to the decrease in CD4+, CD8+ and NK lymphocytes [74]. Interferons (IFN) are a family of cytokines that play a central role in innate immunity to viruses and other microbial pathogens. The IFN-receptor binding induces a cascade of signals with activation of genes coding for proteins with antiviral, antiproliferative or immunomodulatory properties [70][75][70,75]. Normally the interaction between the IFN-γ and IL-6/sIL-6R signals contributes to the recruitment and subsequent clearance of neutrophils, thereby controlling infection and resolution of acute inflammation as well as influencing the transition between innate and adaptive immunity [76]. In patients infected by SARS-CoV-2, a higher IL-6/IFN-γ ratio may be related to an enhanced cytokine storm [74]. These observations suggest that in both SARS patients, and in patients with COVID-19, AKI may have an inflammatory etiology mediated by a cytokine storm. The cytokine storm is associated with an inflammatory process that originates in a local site and spreads via the systemic circulation. The inflammatory process can cause dysfunction in organs, particularly when tissue edema causes an increase in extravascular pressures and a consequent decrease in tissue perfusion. Compensatory repair processes arise soon after the beginning of inflammation, and in a lot of cases they can completely re-establish tissue and organ function. However, when a severe inflammation condition injures local tissue structures, healing occurs with fibrosis, which can cause permanent organ dysfunction [70]. In fact, when a cytokine storm occurs, the immune system may not be able to kill SARS-CoV-2, but it can kill large numbers of normal cells and damage organs [63]. In support of the hypothesis that AKI in COVID-19 patients may be the consequence of inflammatory damage, a cohort study found that the CT scan of kidneys showed a reduced density, indicative of inflammation and edema [8]. In addition to being frequently associated with the cytokine storm, severe lung infections often require prolonged ventilatory support. This predisposes to the development of sepsis, classically defined by marked hypotension which requires treatment with inotropic drugs. Therefore, in patients with COVID-19 or acute respiratory distress syndrome (ARDS), it is plausible that persistent hypotension and vasoconstriction induced by inotropics can participate in the fall of the glomerular filtrate and consequent acute tubular necrosis [77]. In the most recent studies, the hypothesis of a multifactorial etiology of renal damage in COVID-19 is confirmed. Su et al. [66] in a study performed by analyzing autopsy kidney samples showed pigmented casts with high levels of creatine phosphokinase, attributable to rhabdomyolysis. In rhabdomyolysis the massive release of myoglobin due to muscle damage can cause kidney dysfunction. Myoglobin, in fact, shows its renal toxicity through various mechanisms: renal vasoconstriction related to the hyperactivation of RASS by hypoperfusion and the reduction of nitric oxide levels; intratubular cast formation; direct toxicity on renal tubular cells. These processes result in acute tubular necrosis [78]. Rhabdomyolysis in COVID-19 patients is hypothetically multifactorial. In fact, it may be secondary to a direct cytotoxic effect of SARS-CoV-2 on the muscle, tissue hypoxia due to hyperventilation or also to drug-induced damage [66]. Presumably, in COVID-19 patients who develop rhabdomyolysis, it may participate in the pathogenesis of AKI. Su et al. [66] demonstrated the presence of erythrocyte aggregates, without platelets or fibrinoid fragments, which obstructs the lumen of the peritubular and glomerular capillaries in COVID-19 patients. Erythrocyte aggregation, presumably induced by inflammation (reflected by a high rate of erythrocyte sedimentation) and hypotension, can potentiate oxidative stress, inflammation and complement activation, aggravating microvascular damage [64]. Furthermore, occlusion of microvascular lumens by erythrocytes has been associated with a variety of endothelial lesions [66]. Normally, in endothelial cells of the kidney, only ACE is expressed without detectable ACE-2 [79]. Therefore, the renal endothelium cannot be infected directly by SARS-CoV-2. However, this cannot be totally excluded, since ACE-2expressioncan be changed in pathological states or by drugs [66]. Varga et al. recently concluded that SARS-CoV-2 infection induces endothelitis in various organs, directly and indirectly, and that could explain the systemic impairment of microcirculation [80]. Further studies are necessary to better understand the genesis of renal endothelial lesions. Of note, a recent study has proposed a new route for SARS-CoV-2 invasion of host cells via an alternative cell receptor known as CD147 (and also called basigin), which is a transmembrane glycoprotein and is expressed on all endothelial cells [81]. Recently, the high incidence of thromboembolic events in COVID-19 patients suggests that SARS-CoV-2 may play an important role in inducing coagulopathy. Analyzing the hematological profile of COVID-19 patients, a state of hypercoagulability emerged. In fact, high plasma levels of reactive protein C, fibrinogen, D-dimer and ferritin, associated with thrombocytopenia, were found in these patients [71][82][71,82]. Recently, clinical and autopsy reports from China and the U.S. confirm the development of disseminated intravascular coagulation following SARS-CoV-2 infection, with evidence of microangiopathy in several organs. In fact, the activation of macrophages associated with COVID-19, the storm of cytokines and the molecular proteins associated with the damage can cause both tissue factors’ release and the activation of coagulation factors predisposing to hypercoagulability. In some cases, this state of hypercoagulability could favor the evolution of acute tubular necrosis into cortical necrosis and, therefore, the development of irreversible renal damage. These observations suggest that low back pain and microhematuria observed in some positive COVID-19 patients may be manifestations of renal infarction [64]. The SARS-CoV-2 contribution to the development of CKD could involve pathways similar to those described for the acute kidney injury. Indeed, it has been observed that a non-trivial portion of patients develop signs of tubular or glomerular damage during the infection. The direct tubule-glomerular cellular injury, due to the virus, often manifests with proteinuria and hematuria that, in turn, could start a chronic, non-reversible, process [83]. It has been shown that proteinuria exerts a direct toxic effect on renal tubular cells and promotes renal fibrosis over time [84][85][84,85]. In conclusion, the renal damage observed in COVID-19 patients is the result of complex mechanisms induced directly and indirectly by SARS-CoV-2 that predispose to the development of renal dysfunction (Figure 1).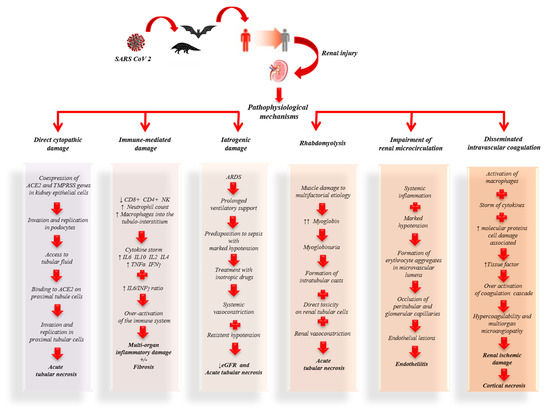
Figure 1.
Pathophysiological mechanisms of kidney damage associated with coronavirus disease 2019 (COVID-19).
6. Histopathology
As mentioned before, the virus gains access to the kidney via the ACE-2receptor for which it uses to enter entrance to target cells [62][86][87][62,86,87] and it has been shown that the ACE-2 receptor of SARS-CoV-2 is highly expressed in renal proximal tubule cells [63][88][89][63,88,89]. Possible mechanisms for kidney injury in COVID-19 include direct infection of the renal parenchyma [47]. Diao et al. [65] retrospectively analyzed the clinical data on renal function from 85 cases of COVID-19; a similar analysis on kidney abnormalities in 26 autopsies of COVID-19 patients was conducted by Su et al. [66]. Using light microscopy, ultrastructural observation and immunostaining have been very informative to understand the extent of kidney injury. Light microscopy examination using Hematoxylin and Eosin staining may show the extent of tubular atrophy and interstitial disease, these are strong histological markers of renal damage and may also predict progression of renal failure. Using light microscopy, proximal tubule injurywas observed with the loss of brush border and vacuolar degeneration with necrosis and detachment of the epithelium observed in the lumen of the tubules; dilatation of the tubular lumen was also noted together with cellular debris resulting from necrosed cells. Erythrocyte aggregates were also observed, causing obstruction of peritubular and glomerular capillary loops, with no obvious erythrocyte or platelet fragmentation or any fibrin thrombi, and less erythrocyte aggregation was observed in peritubular capillaries in those cases with predominantly glomerular loop obstruction [65][66][65,66]. Hemosiderin granules were identified in the renal tubular epithelium of a few patients, while pigmented casts were found in a small number of patients who had high levels of creatine phosphokinase, indicating the presence of rhabdomyolysis. Su et al. noted some cellular swelling of the renal distal tubules and collecting ducts, with some edematous expansion of the interstitial space, but without any significant inflammation. They also noted infiltration of lymphocytesin areas of nonspecific fibrosis, including subcapsular areas. Glomeruli were intact, with various degrees of morphologic changes: nodular mesangial expansion and arteriolar hyalinosis; endothelial cell swelling and degeneration was observed in a small number of COVID-19 patients but these were older individuals with a history of diabetes and hypertension; in other cases podocyte vacuolation and detachment from the glomerular basement membrane was seen; in 2 patients with proteinuria and diabetes, focal segmental glomerulosclerosis was also noted. Immunohis to chemical studies utilizing an anti-SARS-CoV nucleoprotein antibody against the viral nucleocapsid protein (NP) and indirect fluorescence microscopy, showed the NP antigen was present in the renal tubular cells of the infected tissues, suggesting that SARS-CoV-2 can directly infect kidney tubules. Moreover, Diao et al. observed that the presence ofSARS-CoV-2 virus resulted in the infiltration of high levels of CD68+ macrophages into the tubulo-interstitium, and it is feasible that these macrophages could release proinflammatory cytokines which in turn would cause renal tubular damage. In contrast, CD8+ T cells were observed only in moderate numbers in the examined tissues, while CD4+ T cells and CD56+ natural killer (NK) cells were seldom found. Diao et al. also observed the deposition of the C5b-9 complement complex (also known as the membrane attack complex pathway) on the apical brush border of renal tubular epithelial cells, whereas C5b-9 expression was absent in normal kidney tissue. The complex may cause cell death and therefore renal injury and may be an important factor in the pathogenesis of tubulo-interstitial damage [90][91][90,91]. All these findings suggest that SARS-CoV-2 infection causes acute tubular necrosis not only through direct cytotoxicity, but also via immune mediated mechanisms. Observations using transmission electron microscopy (EM) of kidney tissue from autopsies of COVID-19 cases has shown the presence of coronavirus-like particles in the cytoplasm of proximal and distal tubular cells; as well as in podocytes, podocyte foot processes and the glomerular basement membrane. In two autopsy specimens observed by Diao et al., it was noted that the cells in infected kidney tissue were swollen, with enlargement of mitochondria and lysosomes, and viral particles were observed in the broken lysosomes in the cytoplasm. These findings demonstrate SARS-CoV-2 can directly infect kidneys and why patients with COVID-19 often present with proteinuria, hematuria and AKI. As has already been mentioned with the light microscopy observations, EM also showed erythrocytic obstruction of peritubular capillary lumens with injury of the endothelium. Aggregation of erythrocytes in segmental glomerular capillary loops was frequent, but without inflammation or necrosis. Intriguingly, according to a recent study [14] about endothelial cell infection and endothelitis in COVID-19, endothelial dysfunction is probably one of the main causes of microvascular dysfunction by causing vasoconstriction leading to organ ischemia, inflammation with associated tissue edema and a procoagulant state. The principal findings of light microscopy, immunofluorescence and EM are shown in Figure 2.
Figure 2.
Histological features of kidney from COVID-19 patients.