Asthma is a chronic inflammatory disease involving reversible airway obstruction, bronchial hyperreactivity (BHR), mucus overproduction, and airway narrowing and remodeling. The symptoms include coughing, wheezing, and breathlessness and may become aggravated at night or during physical activity, resulting in decreased life quality, impaired productivity, and significant utilization of healthcare resources. Younger individuals are affected more frequently than adults: 9.1% of children, 11.0% of adolescents, and 6.6% of adults experience asthma symptoms, and the prevalence across all age groups is increased in high-income countries. The worldwide mortality rate for childhood asthma reaches up to 0.7 deaths per 100,000, and children who suffer from severe asthma have a greater risk of developing chronic obstructive pulmonary disease (COPD) in adulthood. Genes play a larger role in children’s asthma.
1. ADAM Molecules in General
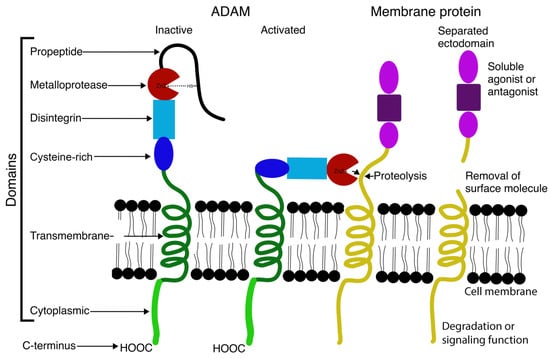
A
disintegrin and metalloproteinase (ADAMs
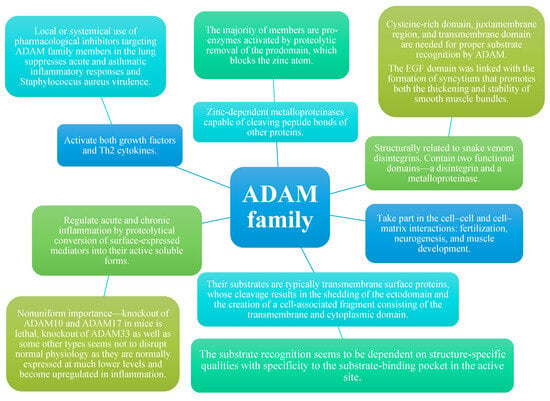
) are types of zinc-dependent metalloproteinases capable of cleaving peptide bonds of other proteins. These membrane-anchored proteins are structurally related to snake venom disintegrins and contain two functional domains—a disintegrin and a metalloproteinase. They take part in many biological processes including the cell–cell and cell–matrix interactions, such as fertilization, neurogenesis, and muscle development
[1][2][6,7]. ADAMs’ substrates are typically transmembrane surface proteins, whose cleavage results in the shedding of the ectodomain and the creation of a cell-associated fragment consisting of the transmembrane and cytoplasmic domain
[3][4]. The substrate recognition seems to be dependent on structure-specific qualities with specificity to the substrate-binding pocket in the active site rather than a defined amino acid motif
[3][4]. Other domains such as the cysteine-rich domain, juxtamembrane region, and transmembrane domain are likely needed for proper substrate recognition by ADAM
[3][4]. The EGF domain in other members of the ADAM family was linked with the formation of syncytium that promotes both the thickening and stability of smooth muscle bundles
[4][8] [
Figure 1].
Figure 1.
Diagram of ADAM domains and function.
ADAMs can regulate acute and chronic inflammation by proteolytical conversion of surface-expressed mediators into their active soluble forms
[3][4]. They can activate both growth factors and Th2 cytokines
[5][9]. Acute and asthmatic inflammatory responses, as well as Staphylococcus aureus virulence, are suppressed with the usage of pharmacological inhibitors that target ADAM family members in the lung either locally or systemically
[3][4]. The majority of ADAMs are proenzymes activated by proteolytic removal of the prodomain, which blocks the zinc atom
[3][4]. The importance of ADAM molecules is not uniform. Although knockout of ADAM10 and ADAM17 in mice is lethal, knockout of
a disintegrin and metalloprotease 33 (ADAM33
) as well as some other types seems not to disrupt normal physiology as they are normally expressed at much lower levels and become upregulated in inflammation
[3][4] [
Figure 2].
Figure 2. Properties of members of ADAM family [1][2][3][4][5]. Properties of members of ADAM family [4,6,7,8,9].
ADAM33 is widely recognized to be a susceptibility gene for asthma with a possible role in the origins of this disease
[5][6][5,9]. The findings of numerous studies carried out since its discovery turned out to be promising, however, partially contradictory among different or even the same races and populations
[7][10]. It is worth saying that ADAM33 alleles were associated with an increased risk of fibrosis, COPD, BHR, accelerated lung function decline across the life course, and impaired early life function
[3][6][8][4,5,11], which emphasizes its importance in lung disease pathogenesis and airway remodeling.
2. Structure
ADAM33 is a type I 120 kD transmembrane protein
[1][9][6,12]. Its gene is located on 20p13 and comprises 21 introns and 22 exons encoding a protein of 813 amino acids
[10][13]. ADAM33 consists of a signal sequence, the NH2-terminal prodomain, and catalytic, metalloprotease-like, disintegrin-like, cysteine-rich, epidermal growth factor-like, transmembrane, and cytoplasmic domains
[2][7], with an mRNA sequence that borders with the 3′-untranslated region (UTR)
[5][9]. A zinc atom bound in the active site is crucial for its proteolytic ability
[3][4].
Northern blot analysis revealed two transcripts of ADAM33: one band at 5.0 kb and a second of lower intensity at 3.5 kb. Northern blot analysis of total cellular and cytoplasmic RNA revealed that the larger transcript was not present in the cytoplasm of bronchial smooth muscle cells, suggesting the occurrence of a splicing process and that the larger message may contain unspliced intronic regions
[11][14]. Regulatory elements located within those regions contain several important SNPs, and it has been suggested that they may control the efficiency and kinetics of splicing
[12][15]. So far, a complex variety of splicing pathways have been described, such as deletion of exon Q, partial deletion of exon R, deletion of whole or of parts of the PRO/MP domains, and/or deletion of other domains (e.g., secretion signal or disintegrin domain)
[12][15]. Alternative spliced forms of ADAM33 were identified in lung-derived cDNA, and one of the variants is predicted to be a soluble form; moreover, it has been implied that there are other ADAM33 species of yet unknown composition
[4][8].
3. Function
ADAM33 is found mainly on cell membranes in airway smooth muscle cells (SMC) and myofibroblasts
[2][7]. It has the ability to regulate angiogenesis, cell differentiation, and proliferation
[2][7]. With its physiologically balanced activity, it plays a protective
[13][16] and regulatory role
[4][8] since it can release growth factors and modify cell surface receptor expression
[4][8]. ADAM33 was found to positively affect cell fusion
[10][13]. Furthermore, its action significantly impacts the activity of adhesion molecules and cytokines through the release of soluble ectodomains, resulting in either loss of function of a cell surface molecule or transduction of an intracellular signal
[3][4]. It impacts proliferation, differentiation, signaling, and apoptosis
[14][17]. Another role of ADAM33 is to keep cell connections intact, thus protecting cells from the penetration of injurious agents
[15][18]. While ADAM33 was previously thought to be expressed only in mesenchymal cells, recent studies have indicated that it may also be expressed in the epithelial cells of patients with severe asthma
[3][16][4,19]. Interestingly, high levels of ADAM33 mRNA were observed in the granulation tissue of a duodenal ulcer, which indicates ADAM33’s possible role in repairing injured tissues
[4][8]. In addition, it was also found in lung leukocytes, the endothelium, and the basal layer of the epithelium
[3][4]. Research has linked ADAM33 to airway remodeling and BHR through the epithelial–mesenchymal trophic unit (EMTU), leading to the proliferation of biosynthetically active fibroblasts, myofibroblasts, and smooth muscles
[17][3]. A loss-of-function mutation of the ADAM33 gene could result in impaired shedding of receptors of growth factors, resulting in increased cellular proliferation. Conversely, a mutation resulting in a gain of function could result in an increase in the shedding of growth factors as well as pro-inflammatory cytokines, causing the type 2 helper lymphocyte-mediated immune response
[13][16]. Indeed, it is suggested that it takes part in the activation of Th2 cytokines
[10][13].
Despite ADAM33 being a membrane protein, its soluble form (sADAM33, 55 kD) is found in high levels in asthmatic airways, which correlates with reduced lung function
[6][5]. A substantial interaction between sADAM33-mediated airway remodeling and sensitivity to allergen exposure, leading to allergic inflammation and BHR in early life, has been found
[6][5]. sADAM33 cleaves membrane domains of stem cell factor c-kit, TNF-related activation-induced cytokines, insulin beta chains, and amyloid precursor proteins
[3][4]. It promotes angiogenesis and myogenesis in vitro
[6][9][5,12] and therefore further impacts airway remodeling independently of inflammation, and its high levels were linked to reduced lung function and the severity of asthma
[5][6][5,9].
A part of ADAM33′s gene containing a single-nucleotide polymorphism (SNP) T_2 encodes the cytoplasmic tail. T_2 allele A, which is known to be associated with asthma exacerbations in adult patients with type 2 inflammatory endotype, may determine vulnerability to shedding and turning into sADAM33
[18][20]. sADAM33′s release is stimulated by transforming growth factor-β2 (TGFβ2)
[18][20]. Maternal allergy during pregnancy triggers the increase in sADAM33 protein and implies the following increase in airway and vascular smooth muscles in utero; however, those changes elicit neither inflammation nor BHR in transgenic mice
[6][5]. Similar changes were observed after the exposure of human embryonic lung explants to sADAM33 in vitro
[6][5]. In the transgenic mice, the remodeling changes induced by sADAM33 did not cause BHR in response to inhaled methacholine and did not increase inflammation
[6][5]. While on its own this remodeling did not impact either BHR or inflammation, the same study proved that the expression of inflammation and remodeling genes, as well as mucous production, was enhanced by sADAM33-expressing mice only after exposure to a common human aeroallergen, house dust mite (HDM) extract. That result was followed by an increase in airway resistance and bronchoalveolar lavage fluid (BALF) eosinophilia
[6][5]. This indicates that sADAM33 increases susceptibility to the Th2 stimulus. The reason behind this could be that the sADAM33-induced airway remodeling causes an increase in the population of fibroblast and smooth muscle cells that are able to produce mediators that intensify inflammatory response
[19][20][21,22]. In addition, the increased angiogenesis caused by sADAM33 could facilitate the migration of eosinophils to lung tissues.
In the other murine model of chronic airway inflammation induced by allergens, there was an observed upregulation of ADAM33 mRNA expression in the lungs
[21][23], and elevated levels of both ADAM33 mRNA and protein were identified in the BALF and bronchial biopsies obtained from individuals diagnosed with asthma
[22][24].
ADAM33’s expression is upregulated during both acute and chronic lung inflammation in patients with COPD and sarcoidosis
[3][4]. Tissue immunoexpression of ADAM33 was higher in viral pneumonia-positive patients than in virus-negative cases
[15][18]. This suggests that ADAM33 plays an important role in the severity and chronicity of the inflammatory process of the lungs and its consequences. Individuals with asthma tend to have an increased expression and enzymatic activity of ADAM33, and this correlates with asthma severity and lung function deterioration, measured in FEV1% (the percentage of the forced expiratory volume in the first second, relative to the total amount of air exhaled during the entire forced vital capacity (FVC) maneuver)
[6][10][23][24][25][5,13,25,26,27]. However, the findings of research on the ADAM33–asthma severity relationship are partly inconsistent since certain investigations on this topic showed no significant association
[26][28].
ADAM33’s increased expression has a potential role in asthma’s eosinophilic/type 2 inflammatory endotype, known to be the most severe and treatment-resistant, which requires the development of novel and nonsteroidal therapeutics
[18][20]. Furthermore, ADAM33 was reported to be associated with a mixed type of eosinophilic/type 2 and neutrophilic inflammations
[18][20]. Its expression was proven by studies to be increased in human fibroblasts in vitro and in asthmatic patients by exposure to interleukin 4 (IL-4) and IL-13 in a time- and concentration-dependent manner
[18][21][20,23]. In mice, its expression was increased also after stimulation with IL-33
[3][4]. Interestingly, the response to exposure to HDM extract was substantially different in ADAM33 knockout mice in comparison to the wild-type mice. In the knockout mice, the expression of Th2-type inflammatory genes Ccl11, Il-5, and Il-13, but not Cxcl1, was suppressed and CCL11, IL-5, and eosinophil levels were reduced in BALF, which indicates ADAM33′s role in the regulation of Th2-type inflammation
[6][5].
Research on epigenetic regulation of ADAM33 showed no difference in the methylation pattern of its promotor between asthmatic and regular fibroblasts.
[27][29]. However, it proved the regulatory hypermethylation of CpG islands within its promotor in the presence of epithelial cells and the corresponding hypomethylation in fibroblasts; therefore, this mechanism probably impacts cell-specific expression.
[27][29]. The repression of ADAM33 in this mechanism appears to be a consistent characteristic of airway epithelial cells, regardless of the presence or absence of asthma
[16][19]. On the other hand, significant differences in methylation patterns in ADAM33 were found between healthy individuals and patients with allergic rhinitis, and hypermethylation of ADAM33 was significantly associated with lower eosinophil counts
[28][30]. TGF-
β2 was found to contribute to chromatin condensation with deacetylation of histone H3, which downregulates ADAM33 expression in both normal and asthmatic fibroblasts
[2][7]. An increase in TGF-
β1 levels in the airways of asthmatic patients both suppresses ADAM33 and promotes its external domain shedding
[2][7].