Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Wei Zheng and Version 2 by Lindsay Dong.
The prediction of three-dimensional (3D) protein structure from amino acid sequences has stood as a significant challenge in computational and structural bioinformatics for decades. TRecently, the widespread integration of artificial intelligence (AI) algorithms has substantially expedited advancements in protein structure prediction, yielding numerous significant milestones. In particular, the end-to-end deep learning method AlphaFold2 has facilitated the rise of structure prediction performance to new heights, regularly competitive with experimental structures in the 14th Critical Assessment of Protein Structure Prediction (CASP14).
- AlphaFold2
- contact map
- deep learning
- distance map
- end-to-end methods
- multi-domain proteins
- protein language model
1. Introduction
Proteins are macromolecules that play important roles in facilitating the essential functions vital for life’s sustenance. Their pivotal involvement spans a diverse array—providing structural support to cells, safeguarding the immune system, catalyzing crucial enzymatic reactions, orchestrating cellular signal transmission, regulating the intricate processes of transcription and translation, and encompassing the synthesis and breakdown of biomolecules. Moreover, they contribute significantly to the regulation of developmental processes, biological pathways, and the constitution of protein complexes and subcellular structures. These diverse and remarkable functions originate from their distinct three-dimensional (3D) structures, which vary across different protein molecules. Since Anfinsen showed that the tertiary structure of a protein is determined by its amino acid sequence in 1973 [1], understanding the protein sequence–structure–function paradigm has emerged as a fundamental cornerstone within modern biomedical studies. Due to significant efforts in genome sequencing over the last few decades [2][3][4][2,3,4], the number of known amino acid sequences deposited in UniProt [5] has grown to over 250 million. Despite the impressive number of data, the amino acid sequences themselves only offer limited insights into the biological functions of individual proteins, as these functions are primarily determined by their three-dimensional structures.
Some of the most widely used experimental techniques for determining protein structures include X-ray crystallography [6], NMR spectroscopy [7], and cryo-electron microscopy [8]. Despite their accuracy, the considerable human involvement and substantial expenses involved in experimentally resolving a protein’s structure have hindered advancement in the number of solved protein structures. Consequently, the expansion in solved protein structures has considerably trailed the accumulation of protein sequences. At present, the Protein Data Bank [9] (PDB) contains structures for approximately 0.21 million proteins, accounting for less than 0.1% of the total sequences cataloged in the UniProt database [10]. This disparity highlights the ever-widening gap between known protein sequences and experimentally solved protein structures. Nevertheless, owing to substantial collective efforts within the scientific community in recent decades [11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][11,12,13,14,15,16,17,18,19,20,21,22,23,24,25], computational approaches have made remarkable progress, through which an increasing fraction of sequences in various organisms have had their tertiary structures reliably modeled [26][27][28][29][30][31][32][33][34][35][36][37][38][39][26,27,28,29,30,31,32,33,34,35,36,37,38,39]. For example, the first version of AlphaFold demonstrated exceptional predictive capabilities in protein structure prediction by employing the deep learning-based distance map prediction during the 13th Critical Assessment of Protein Structure Prediction (CASP13). Furthermore, with the utilization of the end-to-end deep learning approach, the AlphaFold2 has facilitated the rise of structure prediction performance to new heights, regularly competitive with experimental structures in CASP14. These methodologies have significantly contributed to diverse biomedical investigations, including structure-based protein function annotation [40][41][42][43][44][40,41,42,43,44], mutation analysis [45][46][47][48][49][50][51][52][45,46,47,48,49,50,51,52], ligand screening [53][54][55][56][57][58][59][53,54,55,56,57,58,59], and drug discovery [60][61][62][63][64][65][60,61,62,63,64,65].
2. Protein Structure Prediction
2.1. Template-Based Modeling (TBM) Methods
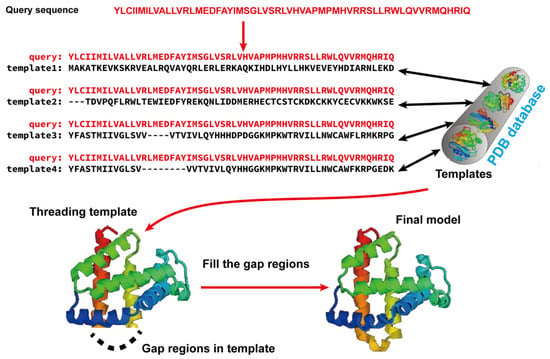
Template-based modeling (TBM) methods have emerged as pivotal approaches in the realm of computational biology for predicting protein structures. TBM leverages known protein structures, referred to as templates, from the PDB to predict the structure of an unknown protein (target), assuming that the target shares a significant degree of sequence similarity with the template. As shown in Figure 12, TBM methods usually consist of the following four steps: (i) identifying templates related to the protein of interest, (ii) aligning the query protein with the templates, (iii) building the initial structural framework by replicating the aligned regions, and (iv) constructing the unaligned regions and refining the structure. TBM can be classified as homology modeling (comparative modeling), which is often employed when there is substantial sequence identity—typically 30% or greater—between the template and the protein of interest, and threading (fold recognition), which is used when the sequence identity drops below the 30% threshold [66].Figure 12. Illustration of template-based modeling (TBM) methods. Starting from a query sequence, templates are identified from Protein Data Bank (PDB) and subsequently aligned with the query protein sequence. Then, the final structural model is constructed by replicating the aligned regions and refining the unaligned regions.
In homology modeling, high-quality templates are detected and aligned using straightforward sequence–sequence alignment algorithms, such as dynamic programming-based techniques like the Needleman–Wunsch [67] algorithm for global alignment and the Smith–Waterman [68] algorithm for local alignment. BLAST [69] is another widely used tool to identify templates and generate alignments, which initially identified short matches between the query and template, and then extended these matches to generate alignments.
In threading, since the sequence identity between the best available template and the query protein falls below 30%, it is hard to identify templates simply based on straightforward sequence–sequence alignment algorithms. Hence, the 1D profile of local structural features is used to represent a template’s 3D structure, because they are often more conserved than the amino acid identities themselves and, thus, can be used to identify and align proteins with similar structures but more distant sequence homology. A commonly used sequence profile is the Position-specific Scoring Matrix (PSSM), which captures the amino acid tendencies at each position within the multiple sequence alignment (MSA). The PSSM is iteratively employed to search through a template database, aiming to identify distantly homologous templates for a specific protein sequence. One popularly used profile-based threading algorithm is MUSTER [70], which combines various sequence and structural information into single-body terms in a dynamic programming search, as follows: (i) sequence profiles; (ii) secondary structures; (iii) structure fragment profiles; (iv) solvent accessibility; (v) dihedral torsion angles; and (vi) hydrophobic scoring matrix. In addition to PSSMs, profile hidden Markov models (HMMs) are another type of sequence profile.
Given the recent substantial improvements in contact and distance map prediction using deep learning, which will be discussed later, threading methods guided by these maps represent the cutting edge in fold recognition, achieving superior accuracy compared to general profile or profile HMM-based threading methods. Among these approaches, EigenTHREADER [71][73] utilized the eigen decomposition of contact maps to derive the primary eigenvectors, which were used for aligning the template and query contact maps. CEthreader [72][74], employing a similar eigen decomposition strategy, outperformed pure contact map-based threading methods by integrating data from local structural feature prediction and sequence-based profiles. map_align [21], on the other hand, introduced an iterative dual dynamic programming technique to align contact maps, while DeepThreader [73][75] leveraged predicted distance maps to establish alignments. Most recently, DisCovER [74][76] integrated deep learning-predicted distance and orientation into the threading method by generating alignments through an iterative double dynamic programming framework.
Furthermore, deep learning-based methods have been directly applied to recognize distant homology templates. The cutting-edge methods, such as ThreaderAI [75][81] and SAdLSA [76][82], conceptualize the task of aligning query sequence with template as the classical pixel classification problem in computer vision, which allows for the integration of a deep residual neural network [77][83] into fold recognition. More recently, the application of language models, originally developed for text classification and generative tasks, to protein sequences marks a significant advancement in the bioinformatics field. Protein language models (PLMs) are a type of neural network with self-supervised training on an extensive number of protein sequences [78][79][84,85]. Once trained, PLMs can be used to rapidly generate high-dimensional embeddings on a per-residue level, which can be viewed as a “semantic meaning” of each amino acid within the context of the full protein sequence. Such representations have proven invaluable in identifying distant homologous relationships between proteins.
Once the templates are identified and aligned with the query proteins, the subsequent step involves building a model by replicating and refining the structure of the template. The most widely used method was MODELLER [16], which constructed tertiary structure models by optimally satisfying spatial constraints extracted from the template alignments, along with other general structural constraints, such as ideal bond lengths, bond angles, and dihedral angles.
With the development of computational techniques, some methods are proposed to convert alignments directly into 3D models. A notable example is I-TASSER [80][81][82][91,92,93], an extension of TASSER [28]. This method utilized a process wherein continuous fragments were extracted from the aligned regions of multiple threading templates identified by LOMETS. These fragments were reassembled during structure assembly simulations. I-TASSER incorporated constraints derived from template alignments and a set of knowledge-based energy terms. These energy terms included hydrogen bonding, secondary structure formation, and side-chain contact formation. The integration of these components was used to guide the Replica Exchange Monte Carlo (REMC) simulation. After clustering low-energy decoys and selecting the centroid of the most favorable cluster, the centroid was compared against the PDB to identify additional templates. The constraints from these new templates, combined with those from the initial cluster model and threading templates, as well as the intrinsic knowledge-based potentials, were employed to direct a subsequent round of structure assembly simulations. The lowest energy structure was selected, which was then subjected to full-atom refinement. Since its first emergence in the CASP7, I-TASSER has consistently achieved top rankings among automated protein structure prediction servers in subsequent CASP experiments [66].
2.2. Fragment Assembly Simulation Methods for Free Modeling (FM)
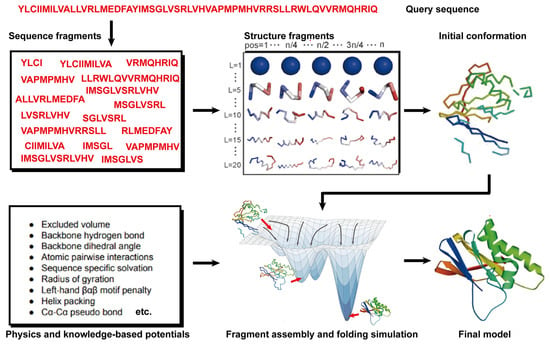
Theoretically, all-atom molecular dynamics (MD) simulations are able to predict protein structures if the computer is powerful enough. However, modern MD simulations can only deal with proteins of less than ~100 amino acids in size. Thus, 90% of the natural proteins cannot be predicted because of the required computational complexity [83][95]. Hence, an alternative method, namely free modeling (FM), was proposed to model protein structures. Compared to MD simulations, FM methods employ the coarse-grained protein elements and physics- or knowledge-based energy functions, together with extensive sampling procedures, to construct protein structure models from scratch. In contrast to TBM methods, they do not depend on global templates. Hence, they are commonly referred to as ab initio or de novo modeling approaches [17][19][17,19]. State-of-the-art FM methods have evolved to assemble protein fragments [84][96]. These fragment assembly techniques assume that protein fragments extracted from the PDB covered most of the conformation of protein folding. Thus, the sampling space was sharply narrowed down. Their implementation involves generating a set of fixed-length (9 residues) and variable-length (15–25 residues) fragments from a repository of known 3D structures (as shown in Figure 23). These fragments are subsequently linked, rotated, and scored to find the global minimum state. This methodology of fragment assembly serves to reduce the exploration of conformational space while ensuring the coherent formation of local structures within the assembled fragments.Figure 23. Illustration of free modeling (FM) methods. Starting from a query sequence, local fragments are identified from databases of solved protein structures, using profile-based threading methods. These fragments are subsequently utilized to construct full-length structural models, guided by physics- or knowledge-based energy potentials.
The first version of Rosetta modeling software, released in 1997, is one of the most well-known FM methods developed by David Baker’s group [17]. Rosetta utilized a three- and nine-residue fragment database for assembly. Particularly, the fragments were selected by quantifying the profile–profile and secondary structure similarity between the query sequence and fragment database within a defined window size. The fragments were simplified to backbone atoms and side-chain centers, and subsequently conducted by simulated annealing Monte Carlo simulations, which exchanged the backbone torsion angles with those of one of the highly scored fragments in the database.
2.3. Contact-Based Protein Structure Prediction
A contact map for a protein of length L is defined as a symmetric, binary L × L matrix. Each element in the matrix represents a binary value, signifying whether the residues form a contact (Cβ-Cβ distance (Cα for glycine) < 8 Å) or not. Since the concept of contact was first brought up, many attempts were made to predict contacts based on correlated mutations in MSAs [85][86][87][97,98,99]. The hypothesis behind these approaches was that residue pairs that are in contact in 3D space would exhibit correlated mutation patterns, also known as co-evolution, because there is evolutionary pressure to conserve the structures of proteins.
n the early 2010s, an increasing number of predictors began integrating deep learning architectures into their prediction methods. A breakthrough occurred in 2017, when Xu’s group introduced RaptorX-Contact [22], which revolutionized contact prediction by integrating deep residual convolutional neural networks (ResNets [77][83]). A Residual Neural Network incorporates an identity map of the input to the output of the convolutional layer, facilitating smoother gradient flow from deeper to shallower layers and enabling training of deep networks with numerous layers. RaptorX-Contact’s utilization of deep ResNets, featuring approximately 60 hidden layers, led to a significant performance leap, outstripping other methods [66]. The introduction of deep ResNets, consisting of approximately 60 hidden layers, enabled RaptorX-Contact to significantly outperform other methods [66].
Due to the latest advances in residue–residue contact prediction, contact-guided protein structure prediction methods have been developed and are becoming more and more successful. The idea of contact-based protein structure prediction methods is described in Figure 34. Starting from a query sequence, an MSA is first generated by searching through databases. The MSA is then used as the input for deep learning methods to predict a contact map. Finally, the contact potential derived from the predicted contact map is used in a folding simulation to predict the final model.
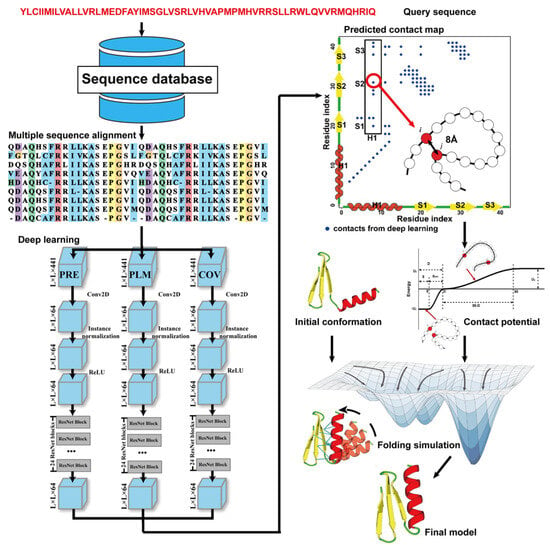
Figure 34. Illustration of contact-based protein structure prediction methods. Starting from a query sequence, an MSA is first generated by searching through databases. The MSA is then used as the input of deep learning methods to predict a contact map. Finally, the contact potential derived from the predicted contact map is used in a folding simulation to predict the final model.
2.4. Distance-Based Protein Structure Prediction
From the definition of contact map prediction, a more detailed extension is distance map prediction. The distinction lies in contact map prediction entailing binary classification, whereas distance map prediction generally estimates the likelihood of the distance between residues falling within various bins (despite attempts made to directly predict real-value distances [88][104]). Distance map prediction gained significant prominence in the field during CASP13 in 2018, when RaptorX-Contact [22], DMPfold [89][105], and AlphaFold [90][106] extended the application of deep ResNets from contact prediction to distance prediction. Among these predictors, AlphaFold, created by Google DeepMind, exhibited superior performance in tertiary structure modeling, as it was ranked as the top one among all groups in CASP13. Leveraging co-evolutionary coupling information extracted from an MSA, AlphaFold employed a deep residual neural network, comprising 220 residual blocks, to predict the distance map for a target sequence, which was subsequently used to assemble protein models. Figure 45 shows the basic steps of distance-based protein structure prediction methods.
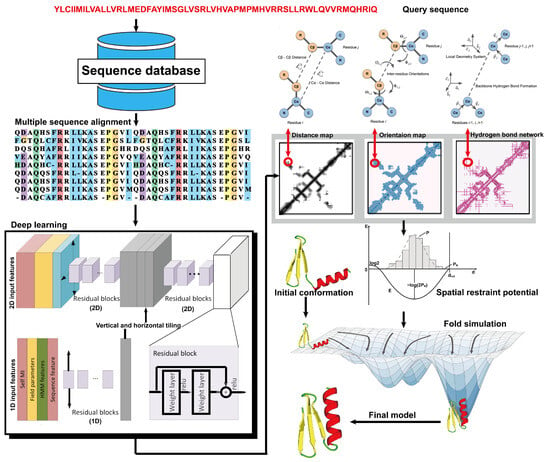
Figure 45. Illustration of distance-based protein structure prediction methods. Starting from a query sequence, an MSA is first generated by searching through databases. Then, the MSA is fed into deep neural networks to predict spatial restraints, such as distance maps, inter-residue orientations, and hydrogen bond networks. Finally, the final structural model is constructed by employing the potentials extracted from the predicted spatial restraints in a folding simulation to identify the lowest energy structure.
2.5. End-to-End Protein Structure Prediction
AlphaFold2 achieved remarkable modeling accuracy and substantially addressed the challenge of predicting the structures of single-domain proteins in CASP14 [91][109]. The success of AlphaFold2 can be attributed, in part, to its unique “end-to-end” learning approach. This end-to-end learning approach eliminates the need for complex folding simulations, allowing deep neural networks, such as 3D equivariant transformers in AlphaFold2, to predict structural models directly.
AlphaFold2 adopted a novel architecture that is quite different from those of previous methods, including the first version of AlphaFold, to accomplish end-to-end structure prediction. The architecture of AlphaFold2 includes the following two primary components: the Trunk Module, which utilizes self-attention transformers to process input data consisting of the query sequence, templates, and MSA; and the Structure (or Head) Module, which employs 3D rigid body frames to directly generate 3D structures from the training components [92][110].
Despite its breakthrough in accuracy and performance, AlphaFold2 has notable limitations, such as increased time consumption with longer protein lengths. To address these challenges, several faster artificial intelligence-driven protein folding tools, based on AlphaFold2, have been developed [93][94][95][111,112,113]. For example, ColabFold [93][111] improved the speed of protein structure prediction by integrating MMseqs2′s efficient homology search (Many-against-Many sequence searching) [96][114] with AlphaFold2 [92][110]. OpenFold [94][112], a trainable and open-source implementation of AlphaFold2 using PyTorch [97][115], achieved enhanced computational efficiency with reduced memory usage, thereby facilitating the prediction of exceedingly long proteins on a single GPU. Similarly, Uni-Fold [95][113] redeveloped AlphaFold2 within the PyTorch framework and reproduced its original training process on a larger set of training data, achieving comparable or superior accuracy and faster speed. Collectively, these developments represent significant strides in enabling rapid and accurate predictions of protein structures.