Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Gabriele Davide Bigoni Ordóñez.
Human papillomaviruses (HPVs) and, specifically, high-risk HPVs (HR-HPVs) are identified as necessary factors in the development of cancer of the lower genital tract, with CaCU standing out as the most prevalent tumor. The mechanisms activated by HR-HPVs during cervical carcinogenesis involve infection by human papillomavirus, cellular tropism, genetic predisposition to uterine cervical cancer (CaCU), viral load, viral physical state, regulation of epigenetic mechanisms, loss of function of the E2 protein, deregulated expression of E6/E7 oncogenes, regulation of host cell protein function, and acquisition of the mesenchymal phenotype.
- HPV
- uterine cervical cancer
- viral load
1. Human Papillomavirus Infection
Human papillomaviruses (HPVs) are small icosahedral viruses, approximately 50 to 60 nm in diameter, non-enveloped, containing a circular double-stranded DNA genome (between 7000 and 8000 bp) (see Figure 1), infecting mucosal and skin epithelia in a specific manner and inducing cell proliferation [5,6,7][1][2][3].
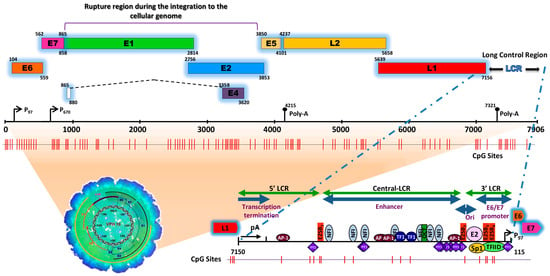
Figure 1. Schematic representation of the structure of the HPV type 16 (HPV16) genome and its long control region (LCR) as a representative model of genital HPVs. The red vertical lines indicate the position of the 112 CpG sites along the viral genome. The bottom of the schematic illustrates the segments into which the LCR is divided as well as the cellular transcription factors that bind to it [8,9,10,11,12,13,14,15][4][5][6][7][8][9][10][11]. To illustrate the genomic structure of HPV16, the latest update of the genomic sequence was used, with NCBI Reference Sequence NC_001526, as well as PISMA software for the localization of each of the CpGs sites [16][12] and Vector NTI® Express Designer Software v1.5.1 (Thermo Fisher Scientific Inc., Waltham, MA, USA) for the identification of the ORFs of each of the HPV16 genes.
The HPV genome is organized similarly to chromatin [17][13] and is divided into three functional regions. The first is a “non-coding upstream regulatory region”, also known as the long control region (LCR) or upper regulatory region (URR). This region contains the p97 core promoter along with cis-enhancer elements that include binding sites for the viral proteins E1 (E1BS) and E2 (E2BS)—required for the commencement of HPV replication—and binding sites for several cellular transcription factors, including Sp1, YY1, TEF-10, AP1, Oct-1, NF1, KRF-1 and glucocorticoid response elements (GREs), required for the initiation of transcription [8,9,10,11,12,13,14,15,18][4][5][6][7][8][9][10][11][14]. The second is called the “early (E) region” and consists of the open reading frames (ORFs) for E1, E2, E4, E5, E6 and E7, where the E1, E2 and E4 proteins are mainly associated with replication, transcription, and viral integration. The E5 protein regulates cell proliferation and apoptosis and facilitates the activity of E6 and E7, while E6 and E7 act as oncoproteins and are associated with cancer development and progression [19,20,21][15][16][17].
2. Cellular Tropism
The uterine cervix is divided into three regions, i.e., the exocervix (also called ectocervix), the endocervix and the squamocolumnar junction (SCJ) or transformation zone (considered a misnomer for a benign process, since the term “transformation” is currently used in oncology to refer to malignant neoplastic transformation). The ectocervix is composed of a non-keratinized stratified squamous epithelium and contains four phenotypically distinct cell populations: epithelial stem or stem-like cells, located in the basal and parabasal layer, and differentiated cells, located in the intermediate and superficial layers. The endocervix is lined by a single layer of mucinous columnar cells (also referred to as columnar epithelium or glandular epithelium). The SCJ is the transition area between the ectocervix and the endocervix and consists of endocervical squamous metaplasia cells, which include endocervical reserve cells (a specialized type of tissue stem cell) and possibly cuboidal cells located, more precisely, in the squamocolumnar junction, which have the capacity to divide and renew [37,38,39,40,41][18][19][20][21][22].
John Doorbar [42][23] extensively described the cellular tropism of HPV. His findings allowed us to establish that in the case of non-keratinized stratified squamous epithelium, the presence of a micro-wound is required that allows infectious virions to access the basal layer and specifically infect stem-like cells. Once infected, the stem-like cells form a reservoir of infection, and in these cells, the viral genome is maintained in an episomal state with a low copy number; as the cells divide, they produce daughter cells that are pushed towards the epithelial surface, giving rise to transient productive infections possibly progressing to high-grade neoplasia or squamous cell carcinoma [42,43,44][23][24][25].
3. Genetic Predisposition to Cervical Cancer
Several genome-wide association studies (GWASs) in different populations have provided evidence that there is a certain genetic susceptibility associated with the development of CaCU. A GWAS study of the British population identified certain single-nucleotide polymorphisms (SNPs) in the PAX8, CLPTM1L and HLA genes, with the SNPs rs10175462 in PAX8, rs27069 in CLPTM1L and rs9272050 in HLA-DQA1 being strongly associated with the risk of developing CaCU [56][26]. Another GWAS study of the Saudi population determined that the SNPs T10C in the GFB1 gene and G399A in the XRCC1 gene were associated with a 1.5-fold increase in the risk of developing CaCU [57][27]. Some studies reported that functional SNPs in codon 72 of TP53 and SNP609 in the NQO1 gene are associated with the risk of developing CaCU [58][28]. Finally, the homozygous CC genotype in the SNP rs4646903 of the CYP1A1 gene—which participates in genetic repair mechanisms—and the CT heterozygous genotype in the SNP rs1801133 of the MTHFR gene—which participates in cellular detoxification—are not only associated with the development of CaCU and high-grade dysplasia, but may also contribute to disease progression [59][29].4. Viral Load
Initially, the detection of HR-HPV viral load was used as an additional test to relate the viral copy number to an active infectious process and reduce false-negative results in HPV diagnostic assays [60][30]. Other studies correlated the viral load of HR-HPV with the age of the patient, histological severity, multiple viral types, the area of the cervical lesion and the sampling method (endocervical and exocervical) [61,62][31][32]. The viral load has also been proposed as a significant marker of progression towards precancerous lesions, that is, as the viral load increases, the risk of cervical lesions increases. The risk is further enhanced if the HPV genotype is high-risk, if the viral infection is persistent during the cervical disease, and if recurrent infections are contracted with different HPV genotypes [63,64,65,66][33][34][35][36].5. Viral Physical State
At the outset of viral infection in the stem-like cells of the ectocervix, the HPV genome persists as a naked nucleic acid (also called an episome), and it depends on the host cell to enable replication. This occurs in the nucleus, with the genome replicating as an extrachromosomal element each time the cell divides [70][37]. As infected cells differentiate and move towards the surface of the epithelium, high levels of viral DNA are replicated, packaged into virions, and released from the surface of the epithelium as virus-laden squamous cells [47][38], thus completing the viral life cycle. By utilizing the Capture-HPV NGS method using tumor biopsies of patients with CaCU, it was determined that HR-HPVs are inserted into intact and repeated regions of the cellular genome, specifically in MYC, NUDT15, MED4, ITM2B, RB1 loci, LPAR6, KLF5, KLF12, PIBF1, RB1, AKT3, SST, ID1, LPP, AFF3, BCL6, CCAT1, CCAT2, RAB11A, RAB22A, MAST4 and MAP2, among others [77][39]. By considering these findings coupled with results from women with normal cytology, those positive for HR-HPV and those with an integrated viral physical status [78,79][40][41] (Figure 2).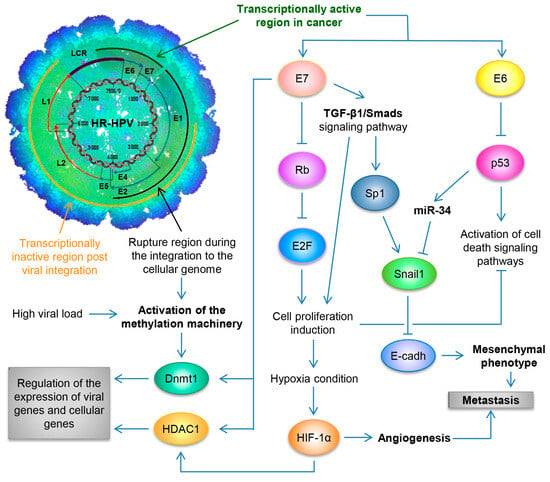
Figure 2. Schematic representation of the molecular mechanisms induced by HPV during the carcinogenic process of the uterine cervix. Both the viral load and the integration phenomenon induce the activation of the methylation machinery, which results in the regulation of the expression of viral genes and cellular genes. Loss of E2 function, either by methylation of the E2SB regions or by deletion of the viral genome during the integration phenomenon, causes the deregulated expression of the E6/E7 oncoproteins, which will consequently induce uncontrolled cell proliferation, evasion of cell death, activation of the angiogenic process and the acquisition of the mesenchymal or metastatic phenotype.
6. Initiation of Epigenetic Mechanisms
6.1. Activation of the Cellular Methylation Machinery
The first studies referring to HPV DNA methylation were carried out in the LCR of the viral genome using different techniques. One of these involved methylation-specific PCR (MSP), which made it possible to report different methylation states (hypomethylated, hemimethylated and hypermethylated) depending on the amplification specificity of the primers [82,83,84,85,86,87][42][43][44][45][46][47].
Based on different publications alluding to the methylation of the LCR of HR-HPVs and given the premise that the methylation machinery is activated as a defense mechanism against foreign genomes, the question is raised as to how HR-HPVs activate the cellular methylation machinery. Based on previous studies, it can be inferred that HPV activates the methylation machinery through two physical mechanisms. The first occurs during HPV infection, when the entry of the viral particles into the target cell activates the methylation of the viral DNA via DNA methyltransferase 1 (Dnmt1).
6.2. Histone Rearrangement
Favre and collaborators were the first to describe that the HPV genome is associated with the canonical histones H2A, H2B, H3 and H4 [17][13]. It was reported that the E2, E6 and E7 proteins of HR-HPV have the capacity not only to bind to the CBP/p300 coactivator complex and inhibit its histone acetyltransferase (HAT) activity, but also to block the ability of p300 to activate p53-responsive promoter elements. This results in the deregulation of cellular signaling, which decreases genome stability and favors the cellular transformation process [105,106,107,108][48][49][50][51].7. Loss of E2 Protein Function
It is generally understood that the oncogenic HPV E2 protein is a negative regulator of the expression of the E6 and E7 oncogenes [72][52]. It is understood that the loss of E2 function can occur in two ways: the first is through the integration process, where breaks in the E1/E2 regions lead to the functional loss of the E2 gene [72,76,109,110][52][53][54][55]; and the second involves the methylation of the CpG sites located in the E2BSs of the HPV LCR, specifically, E2BS1, E2BS3 and E2BS4, which results in the activation of the p97 promoter and the subsequent loss of the repressive function of the E2 protein on the transcription of E7/E6 [96,111,112,113,114][56][57][58][59][60]. Therefore, the loss of E2 function could be considered a key step in carcinogenesis.8. Deregulated Expression of the E6/E7 Oncogenes
Various studies reported that the loss of E2 function—either due to the phenomenon of viral genome integration or due to the methylation of the E2BSs in the HPV LCR—is associated with the overexpression or the aberrant expression of E6 and E7 [102,115,116,117][61][62][63][64]. However, these same studies mentioned that there was no significant difference when comparing the expression levels of the E6/E7 oncogenes in samples that expressed E2 and contained HPV genomes in a purely episomal state or in a coexisting state, with those in samples that contained HPV genomes in a purely integrated state, without E2 expression. This indicates that methylation at specific sites of the E2BSs in the LCR plays an important role not only in the loss of E2 function in those samples harboring transcriptionally active E2 genes, but also in regulating the expression level of E6/E7. This suggests that the overexpression of the E6/E7 oncogenes can be favored only in cases where the following criteria are met: (1) there is a high number of viral genomes in the episomal state with intact E2 genes and with site-specific methylation in E2BS-I and -II; (2) there is a low or moderate viral load, and the viral genomes are integrated at distal sites in a single copy and probably under the control of strong promoter regions in the host cell genome [102,117][61][64].9. Regulation of Host Cell Protein Function
It was reported that the E6 oncoprotein of HR-HPV can evade cell death by apoptosis through two pathways. The first is through the proteasomal degradation of p53 via its association with the ubiquitin ligase UBE3a (E6AP) [118,119,120][65][66][67]. The second is through the interaction of E6 with hADA3—a protein that functions as a coactivator of p53-mediated transactivation for a variety of target promoters—where E6 induces the degradation of hADA3, thus inactivating the function of p53 and overriding the arrest of p14ARF-induced cell growth, despite the presence of normal levels of p53 [121,122][68][69]. Conversely, it is widely accepted that the E7 oncoprotein of HR-HPVs plays two main roles to induce the transforming and proliferative process in cells. Firstly, it binds with members of the retinoblastoma protein (pRb) family, such as p107 and p130 [123][70], which promotes the transcriptional activity of E2F transcription factors, thus regulating cell cycle entry and the progression from the G1 phase to the S phase of the cell cycle [124][71]. Secondly, it destabilizes pRb via degradation through the ubiquitin–proteasome pathway, leading to oncogenic transformation [125][72].10. Acquisition of the Mesenchymal Phenotype
An established feature of solid tumors which are not associated with oncogenic viruses is the acquisition of a mesenchymal phenotype. This is characterized by the overexpression of N-cadherin, vimentin, fibronectin, Twist, FOX C2, SOX 10, MMP-2, MMP-3, MMP-9, Snail and Slug (currently designated as Snai1 and Snai2, respectively, by the HUGO Gene Nomenclature Committee) and a decrease in the expression of E-cadherin (currently designated as CDH1 by the HUGO Gene Nomenclature Committee), desmoplakin, cytokeratin and occludin [203,204][73][74]. Nevertheless, Hellner and collaborators [205][75] reported that both E6 and E7 induced the expression of N-cadherin and that the expression of E7 in primary human foreskin keratinocytes (HFK) induced elevated levels of vimentin and fibronectin, as well as reduced levels of CDH1, while the levels of the regulators Twist and Snai1 remained unchanged. Following the premise that both E6 and E7 of HPV have the ability to induce the expression of, bind to and stimulate the methyltransferase activity of Dnmt1 [185,189][76][77] and that E7 interacts with Mi2β, as well as with HDAC1 and HDAC2, to modulate the expression of cellular genes and viral genes by chromatin rearrangement [157[78][79],213], and knowing that the cell lines HeLa (adenocarcinoma of the cervix), SiHa (squamous cell carcinoma) and Ca Ski (squamous cell carcinoma of the cervix) are representative of the most common types of CaCU with HR-HPV infection and have different viral load and different epithelial origin, a working group previously reported that HR-HPVs can induce a mesenchymal phenotype by negatively regulating the expression of CDH1 through different pathways in which E7, Snai1 and epigenetic mechanisms are involved [214][80].References
- McMurray, H.R.; Nguyen, D.; Westbrook, T.F.; McAnce, D.J. Biology of human papillomaviruses. Int. J. Exp. Pathol. 2001, 82, 15–33.
- WHO. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. In Human Papillomaviruses; WHO: Lyon, France, 1995; Volume 64. Available online: https://www.ncbi.nlm.nih.gov/books/NBK424408 (accessed on 10 August 2023).
- zur Hausen, H.; de Villiers, E.M. Human papillomaviruses. Annu. Rev. Microbiol. 1994, 48, 427–447.
- Apt, D.; Watts, R.M.; Suske, G.; Bernard, H.U. High Sp1/Sp3 ratios in epithelial cells during epithelial differentiation and cellular transformation correlate with the activation of the HPV-16 promoter. Virology 1996, 224, 281–291.
- Butz, K.; Hoppe-Seyler, F. Transcriptional control of human papillomavirus (HPV) oncogene expression: Composition of the HPV type 18 upstream regulatory region. J. Virol. 1993, 67, 6476–6486.
- Gloss, B.; Yeo-Gloss, M.; Meisterenst, M.; Rogge, L.; Winnacker, E.L.; Bernard, H.U. Clusters of nuclear factor I binding sites identify enhancers of several papillomaviruses but alone are not sufficient for enhancer function. Nucleic Acids Res. 1989, 17, 3519–3533.
- Hoppe-Seyler, F.; Butz, K.; zur Hausen, H. Repression of the human papillomavirus type 18 enhancer by the cellular transcription factor Oct-1. J. Virol. 1991, 65, 5613–5618.
- Kanaya, T.; Kyo, S.; Laimins, L.A. The 5′ region of the human papillomavirus type 31 upstream regulatory region acts as an enhancer which augments viral early expression through the action of YY1. Virology 1997, 237, 159–169.
- Kyo, S.; Klumpp, D.J.; Inoue, M.; Kanaya, T.; Laimins, L.A. Expression of AP1 during cellular differentiation determines human papillomavirus E6/E7 expression in stratified epithelial cells. J. Gen. Virol. 1997, 78, 401–411.
- O’Connor, M.; Chan, S.-Y.; Bernard, H.-U. Transcription Factor Binding Sites in the Long Control Region of Genital HPVs. In Human Papillomaviruses; 1995 compendium, part III-A; Los Alamos National Laboratory: Los Alamos, NM, USA, 1995; pp. 21–40. Available online: https://pave.niaid.nih.gov/lanl-archives/compendium/95PDF/3/oconnor.pdf (accessed on 19 August 2023).
- Sailaja, G.; Watts, R.M.; Bernard, H.U. Many different papillomaviruses have low transcriptional activity in spite of strong epithelial specific enhancers. J. Gen. Virol. 1999, 80, 1715–1724.
- Alcantara-Silva, R.; Alvarado-Hermida, M.; Diaz-Contreras, G.; Sanchez-Barrios, M.; Carrera, S.; Galvan, S.C. PISMA: A Visual Representation of Motif Distribution in DNA Sequences. Bioinform. Biol. Insights 2017, 11, 1177932217700907.
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 1977, 21, 1205–1209.
- Parker, J.N.; Zhao, W.; Askins, K.J.; Broker, T.R.; Chow, L.T. Mutational analyses of differentiation-dependent human papillomavirus type 18 enhancer elements in epithelial raft cultures of neonatal foreskin keratinocytes. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 751–762.
- Bhattacharjee, R.; Das, S.S.; Biswal, S.S.; Nath, A.; Das, D.; Basu, A.; Malik, S.; Kumar, L.; Kar, S.; Singh, S.K.; et al. Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol./Hematol. 2022, 174, 103675.
- Harari, A.; Chen, Z.; Burk, R.D. Human papillomavirus genomics: Past, present and future. Curr. Probl. Dermatol. 2014, 45, 1–18.
- Nelson, C.W.; Mirabello, L. Human papillomavirus genomics: Understanding carcinogenicity. Tumour Virus Res. 2023, 15, 200258.
- Fadare, O.; Roma, A.A. Normal Anatomy of the Uterine Cervix. In Atlas of Uterine Pathology. Atlas of Anatomic Pathology; Springer: Cham, Switzerland, 2019; pp. 193–196.
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; McKeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for ‘top down’ differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J. Pathol. 2013, 229, 460–468.
- Kenemans, P.; Davina, J.H.M.; de Haan, R.W.; Hafez, E.S.E. The Cervix. In Atlas of Human Reproduction: By Scanning Electron Microscopy; Springer: Dordrecht, The Netherlands, 1982; pp. 45–54.
- Martens, J.E.; Smedts, F.M.; Ploeger, D.; Helmerhorst, T.J.; Ramaekers, F.C.; Arends, J.W.; Hopman, A.H. Distribution pattern and marker profile show two subpopulations of reserve cells in the endocervical canal. Int. J. Gynecol. Pathol. 2009, 28, 381–388.
- Murall, C.L.; Jackson, R.; Zehbe, I.; Boulle, N.; Segondy, M.; Alizon, S. Epithelial stratification shapes infection dynamics. PLoS Comput. Biol. 2019, 15, e1006646.
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70.
- Griffin, H.; Soneji, Y.; Van Baars, R.; Arora, R.; Jenkins, D.; van de Sandt, M.; Wu, Z.; Quint, W.; Jach, R.; Okon, K.; et al. Stratification of HPV-induced cervical pathology using the virally encoded molecular marker E4 in combination with p16 or MCM. Mod. Pathol. 2015, 28, 977–993.
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222.
- Bowden, S.J.; Bodinier, B.; Kalliala, I.; Zuber, V.; Vuckovic, D.; Doulgeraki, T.; Whitaker, M.D.; Wielscher, M.; Cartwright, R.; Tsilidis, K.K.; et al. Genetic variation in cervical preinvasive and invasive disease: A genome-wide association study. Lancet Oncol. 2021, 22, 548–557.
- Al-Harbi, N.M.; Bin Judia, S.S.; Mishra, K.N.; Shoukri, M.M.; Alsbeih, G.A. Genetic Predisposition to Cervical Cancer and the Association With XRCC1 and TGFB1 Polymorphisms. Int. J. Gynecol. Cancer 2017, 27, 1949–1956.
- Hu, X.; Zhang, Z.; Ma, D.; Huettner, P.C.; Massad, L.S.; Nguyen, L.; Borecki, I.; Rader, J.S. TP53, MDM2, NQO1, and susceptibility to cervical cancer. Cancer Epidemiol. Biomark. Prev. 2010, 19, 755–761.
- von Keyserling, H.; Bergmann, T.; Schuetz, M.; Schiller, U.; Stanke, J.; Hoffmann, C.; Schneider, A.; Lehrach, H.; Dahl, A.; Kaufmann, A.M. Analysis of 4 single-nucleotide polymorphisms in relation to cervical dysplasia and cancer development using a high-throughput ligation-detection reaction procedure. Int. J. Gynecol. Cancer 2011, 21, 1664–1671.
- Schmitt, M.; Depuydt, C.; Benoy, I.; Bogers, J.; Antoine, J.; Pawlita, M.; Arbyn, M. Viral load of high-risk human papillomaviruses as reliable clinical predictor for the presence of cervical lesions. Cancer Epidemiol. Biomark. Prev. 2013, 22, 406–414.
- Lu, X.; Wang, T.; Zhang, Y.; Liu, Y. Analysis of influencing factors of viral load in patients with high-risk human papillomavirus. Virol. J. 2021, 18, 6.
- Veitia, D.; Liuzzi, J.; Avila, M.; Rodriguez, I.; Toro, F.; Correnti, M. Association of viral load and physical status of HPV-16 with survival of patients with head and neck cancer. Ecancermedicalscience 2020, 14, 1082.
- Zhou, Y.; Shi, X.; Liu, J.; Zhang, L. Correlation between human papillomavirus viral load and cervical lesions classification: A review of current research. Front. Med. 2023, 10, 1111269.
- Hortlund, M.; van Mol, T.; Van de Pol, F.; Bogers, J.; Dillner, J. Human papillomavirus load and genotype analysis improves the prediction of invasive cervical cancer. Int. J. Cancer 2021, 149, 684–691.
- Liu, Y.; Xu, C.; Pan, J.; Sun, C.; Zhou, H.; Meng, Y. Significance of the viral load of high-risk HPV in the diagnosis and prediction of cervical lesions: A retrospective study. BMC Women’s Health 2021, 21, 353.
- Ylitalo, N.; Sorensen, P.; Josefsson, A.M.; Magnusson, P.K.; Andersen, P.K.; Ponten, J.; Adami, H.O.; Gyllensten, U.B.; Melbye, M. Consistent high viral load of human papillomavirus 16 and risk of cervical carcinoma in situ: A nested case-control study. Lancet 2000, 355, 2194–2198.
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157.
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23.
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004.
- Canadas, M.P.; Darwich, L.; Sirera, G.; Cirigliano, V.; Bofill, M.; Clotet, B.; Videla, S. New molecular method for the detection of human papillomavirus type 16 integration. Clin. Microbiol. Infect. 2010, 16, 836–842.
- Liu, Y.; Zhang, C.; Gao, W.; Wang, L.; Pan, Y.; Gao, Y.; Lu, Z.; Ke, Y. Genome-wide profiling of the human papillomavirus DNA integration in cervical intraepithelial neoplasia and normal cervical epithelium by HPV capture technology. Sci. Rep. 2016, 6, 35427.
- Kim, J.H.; Choi, Y.D.; Lee, J.S.; Lee, J.H.; Nam, J.H.; Choi, C.; Kweon, S.S.; Fackler, M.J.; Sukumar, S. Quantitative assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Virchows Arch. 2010, 457, 35–42.
- Kim, J.H.; Choi, Y.D.; Lee, J.S.; Lee, J.H.; Nam, J.H.; Choi, C. Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecol. Oncol. 2010, 116, 99–104.
- Turan, T.; Kalantari, M.; Cuschieri, K.; Cubie, H.A.; Skomedal, H.; Bernard, H.U. High-throughput detection of human papillomavirus-18 L1 gene methylation, a candidate biomarker for the progression of cervical neoplasia. Virology 2007, 361, 185–193.
- Turan, T.; Kalantari, M.; Calleja-Macias, I.E.; Cubie, H.A.; Cuschieri, K.; Villa, L.L.; Skomedal, H.; Barrera-Saldana, H.A.; Bernard, H.U. Methylation of the human papillomavirus-18 L1 gene: A biomarker of neoplastic progression? Virology 2006, 349, 175–183.
- Badal, V.; Chuang, L.S.; Tan, E.H.; Badal, S.; Villa, L.L.; Wheeler, C.M.; Li, B.F.; Bernard, H.U. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: Genomic hypomethylation correlates with carcinogenic progression. J. Virol. 2003, 77, 6227–6234.
- Robert, M.F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65.
- Bernat, A.; Avvakumov, N.; Mymryk, J.S.; Banks, L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene 2003, 22, 7871–7881.
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219.
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072.
- Lee, D.; Lee, B.; Kim, J.; Kim, D.W.; Choe, J. cAMP response element-binding protein-binding protein binds to human papillomavirus E2 protein and activates E2-dependent transcription. J. Biol. Chem. 2000, 275, 7045–7051.
- Park, J.S.; Hwang, E.S.; Park, S.N.; Ahn, H.K.; Um, S.J.; Kim, C.J.; Kim, S.J.; Namkoong, S.E. Physical status and expression of HPV genes in cervical cancers. Gynecol. Oncol. 1997, 65, 121–129.
- Kalantari, M.; Blennow, E.; Hagmar, B.; Johansson, B. Physical state of HPV16 and chromosomal mapping of the integrated form in cervical carcinomas. Diagn. Mol. Pathol. 2001, 10, 46–54.
- Cricca, M.; Venturoli, S.; Leo, E.; Costa, S.; Musiani, M.; Zerbini, M. Disruption of HPV 16 E1 and E2 genes in precancerous cervical lesions. J. Virol. Methods 2009, 158, 180–183.
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762.
- Vinokurova, S.; von Knebel Doeberitz, M. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS ONE 2011, 6, e24451.
- Jacquin, E.; Baraquin, A.; Ramanah, R.; Carcopino, X.; Morel, A.; Valmary-Degano, S.; Bravo, I.G.; de Sanjose, S.; Riethmuller, D.; Mougin, C.; et al. Methylation of human papillomavirus Type 16 CpG sites at E2-binding site 1 (E2BS1), E2BS2, and the Sp1-binding site in cervical cancer samples as determined by high-resolution melting analysis-PCR. J. Clin. Microbiol. 2013, 51, 3207–3215.
- Bhattacharjee, B.; Sengupta, S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology 2006, 354, 280–285.
- Thain, A.; Jenkins, O.; Clarke, A.R.; Gaston, K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 1996, 70, 7233–7235.
- Reuschenbach, M.; Huebbers, C.U.; Prigge, E.S.; Bermejo, J.L.; Kalteis, M.S.; Preuss, S.F.; Seuthe, I.M.; Kolligs, J.; Speel, E.J.; Olthof, N.; et al. Methylation status of HPV16 E2-binding sites classifies subtypes of HPV-associated oropharyngeal cancers. Cancer 2015, 121, 1966–1976.
- Fernandez, A.F.; Rosales, C.; Lopez-Nieva, P.; Grana, O.; Ballestar, E.; Ropero, S.; Espada, J.; Melo, S.A.; Lujambio, A.; Fraga, M.F.; et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009, 19, 438–451.
- Zorzi, M.; Del Mistro, A.; Giorgi Rossi, P.; Laurino, L.; Battagello, J.; Lorio, M.; Solda, M.; Martinotti Gabellotti, E.; Maran, M.; Dal Cin, A.; et al. Risk of CIN2 or more severe lesions after negative HPV-mRNA E6/E7 overexpression assay and after negative HPV-DNA test: Concurrent cohorts with a 5-year follow-up. Int. J. Cancer 2020, 146, 3114–3123.
- Giorgi Rossi, P.; Bisanzi, S.; Allia, E.; Mongia, A.; Carozzi, F.; Gillio-Tos, A.; De Marco, L.; Ronco, G.; Gustinucci, D.; Del Mistro, A.; et al. Determinants of Viral Oncogene E6-E7 mRNA Overexpression in a Population-Based Large Sample of Women Infected by High-Risk Human Papillomavirus Types. J. Clin. Microbiol. 2017, 55, 1056–1065.
- Das Ghosh, D.; Bhattacharjee, B.; Sen, S.; Premi, L.; Mukhopadhyay, I.; Chowdhury, R.R.; Roy, S.; Sengupta, S. Some novel insights on HPV16 related cervical cancer pathogenesis based on analyses of LCR methylation, viral load, E7 and E2/E4 expressions. PLoS ONE 2012, 7, e44678.
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135.
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505.
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 1993, 13, 4918–4927.
- Kumar, A.; Zhao, Y.; Meng, G.; Zeng, M.; Srinivasan, S.; Delmolino, L.M.; Gao, Q.; Dimri, G.; Weber, G.F.; Wazer, D.E.; et al. Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol. Cell. Biol. 2002, 22, 5801–5812.
- Shamanin, V.A.; Sekaric, P.; Androphy, E.J. hAda3 degradation by papillomavirus type 16 E6 correlates with abrogation of the p14ARF-p53 pathway and efficient immortalization of human mammary epithelial cells. J. Virol. 2008, 82, 3912–3920.
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105.
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937.
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624.
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773.
- Ribatti, D. Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp. Cell Res. 2017, 353, 1–5.
- Hellner, K.; Mar, J.; Fang, F.; Quackenbush, J.; Munger, K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology 2009, 391, 57–63.
- Au Yeung, C.L.; Tsang, W.P.; Tsang, T.Y.; Co, N.N.; Yau, P.L.; Kwok, T.T. HPV-16 E6 upregulation of DNMT1 through repression of tumor suppressor p53. Oncol. Rep. 2010, 24, 1599–1604.
- Burgers, W.A.; Blanchon, L.; Pradhan, S.; de Launoit, Y.; Kouzarides, T.; Fuks, F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007, 26, 1650–1655.
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458.
- Durzynska, J.; Lesniewicz, K.; Poreba, E. Human papillomaviruses in epigenetic regulations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 36–50.
- Rosendo-Chalma, P.; Antonio-Vejar, V.; Bigoni-Ordonez, G.D.; Patino-Morales, C.C.; Cano-Garcia, A.; Garcia-Carranca, A. CDH1 and SNAI1 are regulated by E7 from human papillomavirus types 16 and 18. Int. J. Oncol. 2020, 57, 301–313.
More