Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Marc Herb.
Reactive oxygen species (ROS) were formerly known as mere byproducts of metabolism with damaging effects on cellular structures. The discovery and description of NADPH oxidases (Nox) as a whole enzyme family that only produce this harmful group of molecules was surprising. Among the Nox isoforms, the NADPH oxidase 3 is the perhaps most underrated Nox enzyme, since it was firstly discovered in the inner ear. Despite the fact that Nox3 is expressed not only in the inner ear but also in various cell types and organs, the “inner ear stigma” remains until today. However ,the involvment of Nox3 is not just limited to the inner ear but extends to various organs and the related diseases.
- NADPH oxidase
- Nox3
- reactive oxygen species
- cardivascular diseases
- hearing loss
- lung diseases
1. Role of Nox3 in Hearing Loss
Hearing loss affects one out of six people and it is one of the major common sensory impairments of humans worldwide [1][2][3]. Hearing loss can be caused by various extrinsic and intrinsic factors, i.e., noise exposure, drug application (including cisplatin), infections and age-related degeneration [3][4][5][6]. The hearing loss in general results from compromised functioning of the organ of Corti in the cochlea and/or the nerve pathways connected to the auditory part of the brain [7]. Several research studies have reported that the nerve connection from the auditory system of the brain to the sound detecting cells (i.e., the hair cells) of the organ of Corti are the most vulnerable parts damaged by endogenous or exogenous sources [8][9][10][11]. The organ of Corti is built up from IHCs and OHCs surrounded by inner and outer phalangeal cells (or Deiters’ cells), inner and outer pillar cells, Hensens cells and Claudius’ cells, all summed up under the term “supporting cells” [12] (Figure 51A). The hair cells detect low- or high-frequency sounds in dependency on their position [13]. Sensory hair cells, in general, do not regenerate in mammals [14][15] and continuous damage results in the permanent loss of hair cells [16]. Oxidative distress is a major driver of hair cell death and subsequent cochlear damage [4][17][18][19][20][21][22][23], which can be induced alongside noise [24][25][26][27], antibiotics [28][29][30], ototoxic anticancer drugs [28][31], infection [32][33] and aging [22][23][34][35][36]. Theses exogenous or endogenous stress factors all result in increased metabolic activity of the cochlea and increased ROS production [26][37][38]. In some cases, like low blood pressure and/or oxygen deprivation, ROS production waves were measured, which started at the luminal surface of the marginal cells in the stria vascularis [39] and re-occurred after reperfusion of the cochlea. The increased ROS levels can last for a long period of time, for example, up to 10 days after noise exposure [37][38][40][41]. This continuous oxidative distress ultimately contributes to death of OHCs and spiral ganglion cells [42][43][44], irreversible cochlea damage and, tragically, permanent hearing loss [8][45][46]. There are many ROS sources in cells with mitochondria [47][48][49] and Nox enzymes as the most prominent ones [50][51][52]. Importantly, mitochondria of OHCs increase their respiratory activity after noise exposure and generate increased amounts of ROS as byproduct [17][53][54], which also contribute to the harmful oxidative damage besides Nox enzymes in general and Nox3 in particular. I point to various excellent reviews about ROS in the inner ear [55][56] or Nox enzymes in this context [22][57] and focus on Nox3-derived ROS. Notably, many studies have used in vivo Wistar rat models, whose hearing ranges are from around 200 Hertz (Hz) to 90 kHz [58] and measured auditory brainstem responses (ABR) for determining the hearing capacity as major experimental output [59][60].
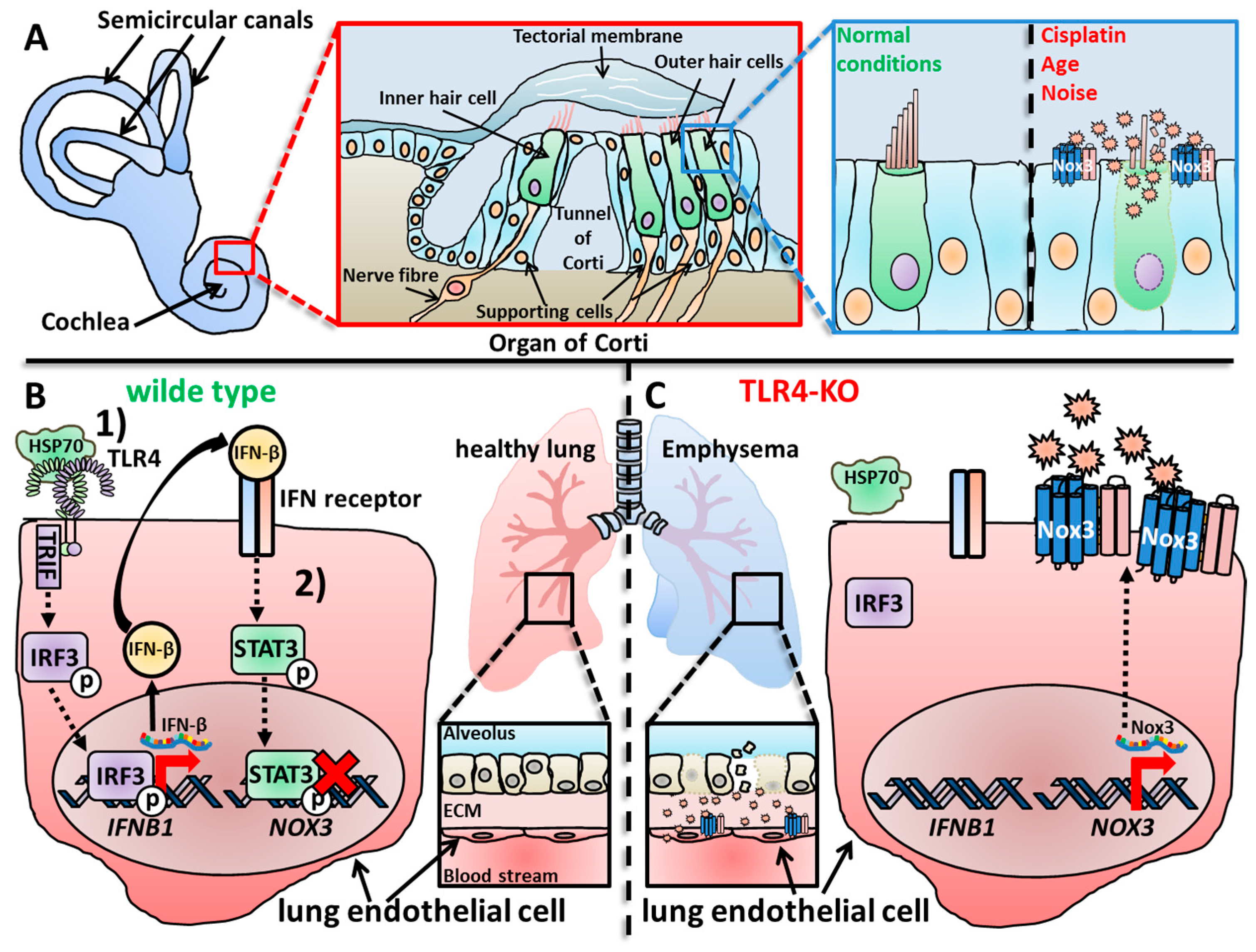
Figure 51. Overproduction of ROS or ROS production in the wrong location can lead to oxidative distress, cellular damage, malfunctioning of tissues and organs and finally manifest in diseases. (A) The cochlea is the organ responsible for hearing and in contrast to the vestibular system, loss of Nox3 leads to a rather protective outcome for the tissue and the hearing capacity. In the cochlea, the organ of Corti is responsible for detection of sound waves and neuronal processing. For that, outer and inner hair cells detect movements of the tectorial membrane, which are induced by incoming sound waves. Under healthy conditions, hair cells and supporting cells function normally; however, under exogenous or endogenous stress conditions, supporting cells up-regulate Nox3. The subsequent ROS overproduction leads to hair cell death and contributes to age-, noise- and drug-induced hearing loss [59][61][62]. (B) In WT mice, the development of lung emphysemas with increasing age is inhibited by a complicated signaling cascade in lung endothelial cells [63][64]. (1) Heat shock protein 70 (Hsp70) activates Toll-like receptor 4 (TLR4)-mediated signaling, which finally leads to activation and translocation of the transcription factor Interferon regulatory factor 3 (IFR3) into the nucleus. IFR3 induces the expression and production of Interferon-β (IFN-β), which is subsequently secreted and (2) activates the lung cells via binding to the IFN receptor in an autocrine manner. The IFN receptor-induced signaling cascade results in activation and translocation of the transcription factor Signal transducer and activator of transcription 3 (STAT3), which then binds to the promoter of the NOX3 gene in result inhibiting the expression of Nox3. (C) In TLR4-deficient animals, this autocrine signaling cascade does not activate, which leads to increased mRNA expression and synthesis of Nox3 and subsequently to an increased ROS production of lung endothelial cells. The accumulating oxidative damage results in destruction of the alveolar structures and subsequently to the development of lung emphysemas in TLR4-deficient mice observed with increasing age.
Nagamani et al. first reported a correlation of four patients with interstitial deletion in the 6q region of the long arm of chromosome 6 and Nox3 expression [65]. Deletions of the 6q region were reported before to be associated with ear anomalies [66][67][68][69], but hearing loss was rarely reported [70]. The study suggested that hearing loss occurred because of interstitial or terminal deletions in the 6q25 region, precisely between the regions 6q25.2 and 6q25.3. This area harbors 12 protein-coding genes, with the NOX3 gene among them. The study described for the first time a possible involvement of Nox3-related inner ear diseases in humans, which started the investigation of Nox3 as harmful ROS source and possible therapeutic target (Section 6) for patients.
1.1. Noise-Induced Ototoxicity
Prolonged exposure to noise is the most common cause of hearing loss worldwide [71][72][73][74][75][76] responsible for 20% of all cases of hearing loss [6]. Exposure to sound pressure levels that exceed 85 dB or immediate exposure to noise impulses lead to irreversible cochlea damage. Exposure to moderate sound levels over a prolonged time period can also harm the spiral ganglion neurons [77][78]. Noise-induced hearing loss is a result from the combined damaging effects of synaptic damage and cochlear hair cell death [79][80]. The noise-induced hearing loss can be temporary or permanent in dependency of the duration, severity and combination of the damaging factors [8]. A number of additional factors can worsen the progress of hearing loss, e.g., other diseases [81][82], social [17][83] and work behavior [84][85] or working conditions [86]. The frequency ranges of the impairment lie between 3.4 and 6 kHz [86]. Previous studies have also suggested genetic components, which might influence the outcome and severity of noise-induced hearing loss [87][88]. For example, mice that already showed age-induced hearing loss were more susceptible to additional noise-induced hearing loss [89]. Furthermore, several mouse lines, which were deficient for antioxidant components, such as superoxide dismutase 1 (SOD1) [38], glutathione peroxidase 1(GPX1) [38], plasma membrane calcium ATPase 2(PMCA2) [90] or Cadherin Related 23 (CDH23) [91] showed also increased sensitivity to noise-induced hearing loss. These findings suggest an important role for ROS in this context in general. Accordingly, a previous study from Ramkumar et al. reported that noise exposure resulted in an increase of ROS levels, oxidative distress and increased pro-inflammatory responses in the chinchilla cochlea [92]. The pro-inflammatory status in the cochlea is mainly attributed to infiltrating immune cells, mainly monocytes [93][94][95], which respond to the cochlear tissue damage and the previously released chemokines from cochlear cells. Together with the already increased ROS production by Nox3 and mitochondria, the pro-inflammatory environment induces a vicious cycle that further increases the cochlear damage instead of dampening it [24][95][96][97]. Importantly, this pro-inflammatory, pro-oxidative setting is not restricted to noise-induced ototoxicity but can be applied to any effect that leads to increased ROS production and cochlear tissue damage. This scenario represents a complex network of cellular mechanisms and communication in the cochlea that still is incompletely understood and needs further investigation [98].
A number of studies performed genetic screens to identify possible factors that might contribute to noise-induced hearing loss. Lavinsky et al. used a well-established Hybrid Mouse Diversity Panel [99][100][101] to investigate possible loci for susceptibility towards noise-induced hearing loss [102]. The Nox3het allele on the murine chromosome 17 was identified as candidate factor. Nox3het mice were exposed to noise and ABR threshold shifts (4, 8, 12, 16, 24 and 32 kHz) were analyzed. The group measured a reduction in the ABR threshold shifts of WT mice in comparison to Nox3het mice at 8 kHz suggesting a role for Nox3 during noise-induced hearing loss in the lower frequency spectrum. Zhao and colleagues performed a genome wide association study (GWAS) in 614 patients of a case-control study to investigate the interplay of noise kurtosis and lifestyle factors with noise-induced hearing loss [103]. Complex noise induces greater damage to the auditory system than steady noise in both animals and humans [104][105]. A complex noise is defined as continuous background noise with temporal appearance of randomly occurring high-level noises [106]. By transforming time-domain variables, like pulse interval distribution or duration, into simple variables by kurtosis [107][108][109], this experimental approach allows to assess the biological effects of complex noise in animal models [104][105][108]. The group reported that the risk of acquiring noise-induced hearing loss was 0.806-times higher for people, which were exposed to complex noise, as shown previously [105][108]. They detected an increased Guanine-to-Tyrosine polymorphism (single nucleotide polymorphism [SNP] rs12195525, GG phenotype) in the locus, which is located in the coding region of the NOX3 gene. They also observed an increased risk for noise-induced hearing loss in GG phenotype patient groups in which further risk factors, such as smoking or high-volume outputs of technical devices, occurred.
The first study that connected the several correlative dots, i.e., Nox3 expression in the cochlea, per se [110], genetic correlations of noise-induced hearing loss with Nox3 [102][103], increased ROS levels in the cochlea as damaging factors [4][17][18][19][22][23], induction of ROS production by noise exposure [26][37][38] and the subsequent hearing loss, was conducted by Mohri and colleagues [59]. The group investigated the role of Nox3 during noise-induced hearing loss in their dtTomato-Cre reporter system for Nox3 detection in mice [59]. The group exposed 2-month-old WT and Nox3-deficient mice (Nox3 marked with the dtTomato fluorescence tag) to harmful noise at 120 dB for three hours and analyzed the ABR thresholds. At day 7, a lower ABR threshold shift at a high frequency (32 kHz) was measured in Nox3-deficient mice in comparison to WT animals. This was accompanied by a reduced OHC loss in Nox3-deficent animals directly linking Nox3 as damaging factor to hearing loss during noise exposure. A recent study from Rousset and colleagues revised previous findings [59][102] concerning the role of Nox3-derived ROS during hearing loss after white noise exposure [62]. Rousset et al. used the previously described C57BL/6J-NOX3het−4J mouse strain [111], which carries a loss-of-function allele of Nox3. They applied RNAscope in situ hybridization on murine cochlea explants and detected strong Nox3 mRNA expression in the spiral ganglion, while Nox3 was only weakly expressed in the stria vascularis and not detectable in the organ of Corti. The latter is contradictory to several previous studies [59][110][112][113]. Additionally, they detected Nox3 mRNA in the peripheral auditory neurons in Rosenthal’s canal. After noise exposure, Nox3 mRNA expression was increased in cochlear explants, precisely in the medial and the apical cochlea turns. The group also analyzed the hearing capacities of Nox3-deficient mice and observed no difference in the audiograms in comparison to WT animals after 6 weeks of age confirming not a general deficit of hearing in Nox3-deficient animals. Deafening noise exposure (116 dB) led to an elevation of hearing thresholds at frequencies between 16 and 32 kHz after 24 h in WT mice. A protective effect in Nox3-deficient animals was only observed for 32 kHz. After 7 days of noise exposure, ABR measurements showed a better recovery of hearing in Nox3-deficient mice, while WT animals showed no recovery. Histological examinations of cochlear explants further showed that Nox3-deficient animals had reduced hair cells loss, conserved auditory synapses and intact neuron integrity, which all were deceased in WT animals. This study nicely confirmed previous results [8][59][61][114][115], showing that Nox3 has no direct role for cochlear development and structures in sharp contrast to the otoconia formation in the vestibular system [116][117][118]. Even worse, after noise exposure, Nox3-mediated ROS overproduction results in increased oxidative distress and damage of cochlear structures (Figure 51A).
Goodarzi and colleagues investigated the combined effects of noise exposure and silver nanoparticles (Ag-NPs) on the cochlear function in rats [119]. The influence, either beneficial or detrimental, of nanoparticles, in general, on biological functions of the organism is a swiftly expanding research topic [98][120][121][122]. However, metallic nanoparticles, in particular, exploit toxic effects on cells by increasing the ROS production and pro-inflammatory cytokine release [123]. Ag-NPs can enter the body in various ways, e.g., via ingestion, inhalation or even skin contact [124]. Previous studies have reported toxic effects of Ag-NPs to the cochlea [125][126][127]. Goodarzi et al. compared completely untreated Wistar rats with rats exposed to loud noise (104 dB) for different time intervals. The animals either received not further treatment or were intra-peritoneally injected with Ag-NPs (100 mg/kg body weight). The group measured distortion product otoacoustic emissions (DPOAEs) for screening the inner ear function [128][129]. Animals showed a higher rate of hearing loss when exposed to both noise and Ag-NP at frequencies of 7.26, 8.47 and 9.86 kHz. Oppositely, malondialdehyde (MDA) and SOD levels in the serum were either increased by noise exposure or Ag-NP treatment alone but were not further increased by the combined treatment. qRT-PCR analysis further showed that TNFSF2, IL6 and NOX3 gene expressions in the cochlea were increased by one of the treatments alone but were not further increased by the combinatory treatment. Further investigations concerning Nox3-derived ROS were not made. A similar research topic was investigated by Shahtaheri et al. The group investigated the effects of white noise in combination with aluminum oxide (Al2-O3) nanoparticles (AO-NPs) on the cochlear structure in rats [130]. AO-NPs are widely used as thermal insulation material [131], and the exposure to workers that are involved in AO-NPs manufacturing [132][133][134] is correlated with many harmful effects on workers’ health [135][136][137]. Additionally, workers are often exposed to extreme noise levels. Regarding this harmful work environment, Shahtaheri and colleagues analyzed the combinatory harmful effects of AO-NPs and noise exposure (95 dB/20 Hz–20 kHz, 8 h per day) on the cochlea of Wistar rats. The group detected reduced auditory capacities analyzed by DOPAE measurements [76][138] and cochlear damage by histochemical analysis in rats exposed to noise. The damage was further increased by treatment with AO-NPs. AO-NP treatment alone did not alter the investigated parameters. Notably, Nox3 mRNA levels also increased after noise exposure in the cochlea, while AO-NP treatment alone did not change the mRNA expression levels of Nox3. The combinatory effect of both increased Nox3 mRNA expression significantly in comparison to noise exposure alone. This was accompanied by OHC and a supporting cell decrease, while IHC numbers showed no alterations. The authors suggested an enhanced damaging effect of white noise exposure and AO-NP treatment on the cochlea due to increased Nox3-mediated oxidative distress. Critically, neither Nox3 knock-down experiments nor ROS measurements were performed in this context. Hence, again direct evidence for a Nox3 involvement is missing in this study.
1.2. Cisplatin-Induced Ototoxicity
Cisplatin is a commonly used chemotherapeutical agent against solid tumors [139][140][141][142][143]. Similar to most chemotherapeutical applications, cisplatin treatment results in strong side effects for the patients like nephrotoxicity and ototoxicity [83][144][145][146][147][148]. Cisplatin-induced nephrotoxicity can be treated with diuretics [149][150], while cisplatin-induced ototoxicity is a much more severe, cumulative and untreatable problem [18][147][151]. It manifests as sensorineural, irreversible hearing loss [152][153][154][155][156] due to damage of the organ of Corti in the cochlea [4][157][158][159]. Specifically, cell death of IHCs and OHCs [83][160][161][162], of spiral ganglion cells [163][164][165][166] and of marginal cells of the stria vascularis [167][168] is increased after cisplatin treatment. Inflammation after cisplatin treatment is another driving factor, which further progresses the cochlear damage [169][170][171][172][173][174]. On the sub-cellular level, cisplatin-mediated cytotoxicity induces DNA damage [175][176], mitochondrial dysfunction [177][178] and increased ROS production by various ROS sources [40][110][178][179][180][181][182]. The accumulating damage due to the oxidative distress further progresses the dysfunction of cochlea [28][160] and vestibular system [182][183][184].
Banfi and colleagues first reported cisplatin-induced Nox3-mediated ROS production by using a co-expression system in HEK293 cells [110]. Mukrerhajea et al. provided further evidence in vivo in the rat cochlea and in vitro in the OHC line UB-OC-1 [185]. Cisplatin treatment induced in both systems increased Nox3 expression and ROS production [186]. Kim and colleagues investigated the role of Nox enzymes during cisplatin-induced ototoxicity in general [187]. They used the mouse auditory cancer cell line HEI-OC1 and in vivo experiments for this approach. Cisplatin treatment induced Nox1 and Nox4 mRNA expression starting after 1 hour. Unfortunately, they claimed that Nox3 mRNA was not detectable; however, the data were not shown in the publication. Notably, in vivo injection of cisplatin for 4 days showed a strong induction of the already basally expressed Nox3 mRNA in the cochlea. However, the group focused on Nox1 and Nox4, and Nox3 as ROS source was not further analyzed. Mohri and colleagues investigated, besides several other important Nox3-related topics, also the role of Nox3 during cisplatin-induced hearing loss [61][188]. The group used their well-established reporter system with the dtTomato-coupled Nox3 protein [59]. Tone-burst stimuli (8, 16, 24 and 32 kHz) were applied on 2-month-old WT and Nox3-deficient mice either treated with cisplatin or left untreated. ABR threshold shifts were measured, and WT animals showed deteriorated ABR thresholds at frequencies of 24 and 32 kHz after cisplatin treatment compared to Nox3-deficient animals, which showed no deterioration. WT mice also showed OHC loss, which was lower in Nox3-deficient mice. TdT-mediated dUTP-biotin nick end labeling (TUNEL) assays confirmed increased apoptosis of OHC in WT animals in this context as reported before [61][189]. In Nox3-deficient animals fewer TUNEL-positive OHC were detected. In the lateral wall of the cochlea and the stria vascularis no TUNEL-positive cells were seen in both WT and Nox3-deficient mice. The group furthermore showed that cisplatin treatment increased Nox3 expression in the cochlea predominantly at the basal turn and in the supporting cells. In detail, no Nox3-expressing OHC either with or without cisplatin treatment could be detected in WT animals, while weak Nox3-expression in IHCs and strong Nox3 expression in supporting cells could be observed at least after cisplatin treatment. Together, these studies provide solid evidence that cisplatin treatment increases the presence of Nox3 in the cochlea, which leads to a harmful elevation of ROS production and finally to ototoxicity. Interestingly, in vivo Nox3 is mainly present in the supporting cells and not the OHCs, which nevertheless suffer the greatest damage through the increased ROS production (Figure 51A).
Several studies have provided evidence for a protective role of the activating adenosine A1 receptors (A1ARs) [190] and its agonist adenosine during cochlea-related diseases [113][191][192][193][194][195][196][197][198]. In this context, Kaur and colleagues investigated the role of ROS for the A1AR signaling during cisplatin-induced ototoxicity [115]. They reported that activation of the A1AR signaling pathway by N6-R-phenylisopropyladenosine (R-PIA) prevents hearing loss induced by cisplatin and OHC damage in the rat in vivo. They used the OHC line UB-OC-1 to investigate a role for Nox3-derived ROS in vitro, since ROS have a pro-inflammatory effect during cisplatin-induced ototoxicity [110][186]. Cisplatin treatment for 24 h induced A1AR mRNA and protein expression and increased Nox3 mRNA as well as the total cellular ROS levels. Treatment with R-PIA reduced ROS generation and Nox3 mRNA expression in UB-OC1 cells and in the rat cochlea. Cisplatin treatment of UB-OC-1 cells also induced phosphorylation and nuclear translocation of Signal transducer and activator of transcription 1 (STAT1), which could be inhibited by additional R-PIA treatment. STAT1 signaling contributes to the pro-inflammatory response during cisplatin-induced ototoxicity [199]. Accordingly, treatment with R-PIA reduced cisplatin-induced expression of TNF in the rat cochlea. However, no experiments after Nox3 knock-down or knock-out were performed. Therefore, evidence for the identification of Nox3 as relevant ROS source in this context is missing.
1.3. Cytomegalovirus-Induced Hearing Loss
Congenital Cytomegalovirus (CMV) infection often leads to sensorineural hearing loss accompanied by neurological and developmental disabilities [200][201][202][203]. Several studies have monitored apoptotic cell death in the murine cochlea [204] in neonatal mice after CMV infection, subsequently leading to sensorineural hearing loss [205][206]. A correlative increase in total cellular ROS levels was also described in this setting [205]. Due to these previous observations, Zhuang and colleagues picked this topic up and investigated the possible ROS sources and the effect of the anti-inflammatory substance Berberine [207] during CMV-induced ototoxicity [208]. The group detected an increase in apoptosis and total cellular ROS in neonatal murine ex vivo cultured spiral ganglion cells. An increase in Nox3 protein expression was also observed. Additional treatment of Berberine reduced apoptosis, ROS levels and Nox3 expression. However, no genetic evidence was given to validate Nox3 as ROS source. Most critically, the authors claimed that Nox3 was connected to mitochondrial ROS production. No specific mitochondrial ROS measurements were performed, and no co-localization studies of Nox3 with mitochondria, e.g., by immunolabeling and fluorescence microscopy, were conducted. Nevertheless, this is so far the one and only study that has described an induction of Nox3 protein expression as response to infection.
1.4. Age-Induced Hearing Loss
Age-induced hearing loss (presbycusis) [209] affects, as the name implies, elderly people. This disease is associated with tremendous social consequences [210][211][212][213]. Similar to other causes for hearing loss, age-induced hearing loss can further progress due to prolonged noise exposure or ototoxic drugs [214]. On the cellular level, the loss of hair cells, spiral ganglion cells and cells of the stria vascularis leads to hearing loss majorly at higher frequencies [211][215]. An increase of age is also accompanied with a disturbance of redox homeostasis not only in the cochlea [22][34], but also in other organs, since gene expression of anti-oxidant systems decrease with age [23][35][36].
Du and colleagues investigated the effects of a HFD in combination with a D-galactosidase-induced rat animal model of aging [216][217] to investigate the cumulative effects on hearing loss [218]. In this animal model, the continuous administration of D-galactose leads to numerous detrimental effects based on metabolic disturbance that mimic the aging process [216][219][220][221]. These effects include dysfunctional mitochondria [220][222][223], increased apoptosis [224][225], neurotoxicity [220][226][227], a shortened lifespan [228] and, after 8 weeks of treatment, symptoms that mimic aging of the cochlea due to increased ROS production [229][230][231][232]. Furthermore, after 8 weeks of D-galactose treatment deletions in the mitochondrial DNA (mtDNA) in the cochlea increase and mitochondria show an oval round shape indicating massive damage. The isolated mtDNA from rat cochlea cells showed increased oxidative damage and subsequent common deletion, which are both biomarkers for oxidative distress, aging and age-related hearing loss [216][233][234][235][236][237]. Du et al. analyzed ABR thresholds and detected the highest ABR threshold shifts for four tested frequencies (4, 8, 16, 32 kHz) in groups treated with both HFD and D-galactose after 12 months. After sole D-galactose treatment Nox3 protein levels increased in the stria vascularis and the spiral ganglion. HFD treatment alone increased Nox3 protein levels only in the stria vascularis. The combined treatment of D-galactose and HFD led to the highest Nox3 expression not only in the stria vascularis and the spiral ganglion, but also in the organ of Corti. Apoptotic cell death in the inner ear was observed for all three conditions, but again the highest cell death rate was reported after the combined treatments. Additionally, all three treatments increased the accumulation of mitochondrial common deletion [238][239], which accompanies mitochondrial damage due to aging [240][241]. Du and colleagues deepened their findings from this previous study [218] with the same D-galactose-induced aging model via RT-PCR and Western blot analysis and reported an increase in Nox3 and p22phox mRNA and protein expression in D-galactose-treated rats in the cochlea [112]. Additional Western blot analysis and TUNEL staining showed that apoptosis increased in the cochlea after D-galactose treatment. These two studies by Du and colleagues gave the first correlative insights of increased Nox3 expression during aging, an associated damaging effect to cochlear structures and the subsequent hearing loss. However, since mitochondria are heavily damaged during this aging model and neither in vivo experiments with Nox3-deficient animals nor in vitro experiments with Nox3 knock-down in cells were conducted, the explicit role and the contribution of Nox3-derived ROS in comparison to ROS produced by the damaged mitochondria remained elusive.
Rousset and colleagues used the A/J mouse strain nmf333, which carries a missense mutation in the p22phox subunit [242], to characterize the role of Nox enzymes in the cochlea during age-induced hearing loss [8]. The group firstly defined age-induced hearing loss in WT animals in their experimental setting. They analyzed ABR threshold levels over an age range from 4 to 26 weeks and observed threshold shifts close to 45 dB after 4 weeks, which progressed up to 75 dB with age. They also detected a progressive hearing loss 32 kHz (in 4-week-old mice) and 5.7 kHz (in 26-week-old mice). In accordance with these data sets, a progressive degeneration of the sensory epithelium from the base to the apical turn was described with a more pronounced cellular degeneration in the basal region. Further analysis of IHC innervation revealed a dramatic decrease in the number of synaptic ribbons per IHC, as well as a decrease in the total neuronal density in the spiral ganglion, which also progressed with age. Since a deficiency of p22phox affects Nox1-4, the group analyzed the presence and distribution of Nox mRNA expression in both the mouse and, highly notably, in the human cochlea. qRT-PCR and in RNAscope in situ hybridization measurements showed high mRNA expression of Nox2, Nox3 and Nox4 in mouse and human cochlea tissue. While Nox2 and Nox4 mRNA was evenly distributed throughout the whole cochlea, Nox3 mRNA was concentrated in the spiral ganglion and moderately expressed in the stria vascularis. Most interestingly, Nox3 mRNA was not detected in hair cells, which is in line with the study from Mohri et al. [59]. p22phox-deficient animals showed no disturbance in hearing at young age in comparison to WT mice. However, the loss of the hearing capacities at high frequencies observed in aged WT mice, was nearly absent in p22phox-deficient animals together with an intact sensory epithelium and preserved synaptic ribbons. The group further performed a transcriptome analysis of 6-week-old cochlea tissue and detected a down-regulation of ryanodine receptors (Ryr) 1, 2 and 3, which are important for Ca2+ homeostasis and accordingly for proper neuronal signaling. Several other genes, all revolving around Ca2+ homoeostasis, such as Otoferlin, Vamp1, and Snap25 or the glutamate transport, such as Slc17a6, Slc17a8 and Gria2 were down-regulated in absence of p22phox. The group narrowed down the auditory neurons as main cell type where the down-regulation was observed. This remarkable study firstly analyzed the mRNA expression of Nox3 in the human cochlea and clearly solidified a rather detrimental effect of Nox presence on cochlear structures, precisely the neuronal part. Unfortunately, like in other previous studies of the Nox3 research field, the group did not clarify the exact interplay of Nox-derived ROS during Ca2+ signaling and the subsequent age-related hearing loss. Moreover, while nicely showing that also Nox2 and Nox4 mRNA is present in the cochlea, the analysis of p22phox-deficient animals only enabled suggestions considering the general role of Nox enzymes in the cochlea and not specifically the role of Nox3, especially since Nox2 and Nox4 might also play important roles in this organ [243][244][245]. Protein expression, for example, in cochlea tissue lysates, was not analyzed, Instead, the research group solely relied on mRNA-detecting techniques. Since the opinion that mRNA always correlates with protein presence or even activity of the protein is outdated [246][247][248][249][250], protein level analysis of the cochlea, especially from human samples would have been a ground-breaking contribution to the field of Nox3-related research. Human-related data sets of this topic are still largely missing to date. In their favor, the group mentioned and discussed these critical points already in their paper. In summary, the studies of Rousset and colleagues [19][62][251], together with Mohri et al. [59] represent milestone research articles considering Nox3 investigations in the inner ear. Continuing in this sense, Mohri and colleagues also investigated the topic of age-induced hearing loss with their generated mouse line, which expresses the fluorescent reporter dtTomato in cells that display Nox3 expression [59]. The group compared the ABR threshold shifts in WT and Nox3-deficient animals after 1, 2 and 6 months after birth. An increase of Nox3 protein in the cochlea as well as increased ABR threshold shifts at frequencies of 8, 24, and 32 kHz occurred in WT mice over time. Nox3-deficient mice showed no ABR threshold shift increase at all. Especially at high frequencies (24 and 32 kHz), the ABR thresholds were higher in WT mice in comparison to Nox3-deficient animals at 6 months from birth. In addition, histologic analysis of the organ of Corti showed that WT mice at 6 months after birth exhibited OHC loss, while hair cell loss in Nox3-deficient mice was significantly lower. These findings suggest that increased Nox3 expression in the organ of Cori leads to OHC destruction and subsequently contributes to age-related hearing loss (Figure 51A).
2. Role of Nox3 during Vertigo
The only study which investigated a rather harmful effect of Nox3-derived ROS on the vestibular system (in contrast to the crucial function of otoconia formation), was conducted by Zhang et al., who investigated factors that influence benign paroxysmal positional vertigo (BPPV) [252]. BPPV is the most common peripheral vertigo-related disease [253][254] occurring in 2.4% of people [255], which increases with age [256]. BPPV is characterized by the detachment of otolith particles, particle movement into the semicircular canal and subsequent loss of otoconial function [257][258]. BPPV is therefore also termed otolithiasis. In dependency how the proper function of the otoconia is impaired, BPPV can be classified in primary BBPV and secondary BPPV. Primary BBPV is induced by factors that directly damage the otoliths or their surroundings, e.g. hair cell damage or loss, endolymph ion changes, decreased otolith protein secretion and defects in otolith-anchoring proteins [257][259]. Secondary BBPV is defined as damage, which is induced as side effect of other harmful events, such as ear surgery, trauma, ototoxic drugs, Meniere’s disease [260] or vestibular neuronitis [261]. Systemic factors like osteoporosis [262], vitamin D deficiency, hypertension, diabetes or cerebrovascular diseases [263] can also contribute to the severity of this disease. Zhang and colleagues focused on vitamin D deficiency during BPPV, since vitamin D is important for proper Ca2+ homoeostasis in general [264][265] and for proper otolith formation and function in particular [261]. Overall, 48 patients with diagnosed BPPV and 48 control patients from the Affiliated Hospital of Inner Mongolia Medical University [266] were analyzed in this study. While no difference in age, body mass index, sex, occurrence of diabetes or hypertension was observed between the groups, BBPV-diagnosed patients showed a decreased bone density and plasma vitamin D levels. Notably, mRNA and protein levels of both OC-90 and Nox3 in the serum were decreased in patients with BPPV. To further analyze the role of vitamin D in this context, vitamin D receptor (VDR)-deficient mice were analyzed. In whole-tissue lysates of the inner ear, mRNA and protein levels of OC-90 and Nox3 were decreased in VDR-deficient mice suggesting a regulatory role of vitamin D in this context. A direct mechanism for VDR-mediated signaling for Nox3-derived ROS production and OC-90 assembly was not investigated.
3. Role of Nox3 during Lung Diseases
For a long time, Nox3-related research only focused on either the inner ear or studies focused on broad expression studies to improve the catalogue, which lists if, when and where Nox isoforms are expressed. Most of the latter studies have not focused explicitly on Nox3, but rather described its expression as additional finding. Zhang and colleagues investigated, for the first time, a possible connection between Nox3 and pulmonary emphysema, which is a major contributor to chronic pulmonary diseases [267][268] in a mouse model [64]. They described developing emphysemas in naive TLR4- and MyD88-deficient mice beginning at 3 months after birth and peaking between 6 months and 1 year. This was reflected by increased lung volumes, enlarged air spaces distal to the terminal bronchioles and by destruction of the normal alveolar architecture. These factors are typical for emphysema [267] and occurred in both knock-out animal strains. Notably, all mice strains did not show any significant differences in any pro-inflammatory parameter that was analyzed. However, TLR4-deficient animals showed a decreased elastase inhibitory capacity and increased elastolytic activity in the lung tissue. Since increased oxidative distress is an important correlative factor of emphysema [269] and lung injuries [270][271][272][273][274][275][276][277], the group analyzed the total antioxidant capacity, namely levels of glutathione (GSH) and other antioxidant components in the branchio-alveolar fluid. A strong decrease of GSH levels was detected in the fluid of knock-out animals. Moreover, isolated lungs and isolated lung cells from TLR4-deficient animals showed increased O2− production in comparison to WT animals. The increased ROS levels further led to more oxidative DNA damage, which is also correlated with emphysema [278]. Interestingly, while Nox3 mRNA was only weakly expressed in WT animal lungs and isolated endothelial lung cells, TLR4-deficient lung samples and lung cells showed an increased Nox3 mRNA expression. Additionally, isolated lung cells from TLR4-deficient animals showed an increased elastolytic activity similar to the lung tissue. Knock-down of Nox3 via siRNA in TLR4-deficient lung cells led to a rescue effect of elastolytic activity, nicely confirming the involvement of Nox3. These results clearly demonstrated the connection of TLR4 deficiency, increased Nox3 expression, Nox3 as cause for the increased elastolytic activity and therefore the developed emphysema. A direct mechanism for Nox3-derived ROS was not investigated at that time. Nevertheless, the study of Zhang and colleagues broke the ”inner ear” stigma of Nox3 in terms of disease developement.
In a follow-up study from Zhang and colleagues, a role of Nox enzymes during hyperoxia was investigated. Hyperoxia can occur during sustained oxygen supply in critically ill patients, which can result in respiratory failure [279][280]. Hyperoxia is also an established model for oxidant-induced lung injury [281][282]. Previous reports of the group demonstrated that TLR4-deficient mice showed increased oxidant production in lung tissue and subsequent lung destruction [64], as well as enhanced susceptibility to hyperoxia-induced acute lung injury [283]. An increase in Nox3 mRNA was also reported in TLR4-deficient animals, and siRNA-mediated knock-down partially rescued the phenotype related to TLR4-deficiency [64]. WT mice exposed to hyperoxia showed increased TLR4 mRNA and protein levels in mouse lung endothelial cells and lung lysates. TLR4-deficient mice were more susceptible to hyperoxia, as reported before [283], but interestingly, Nox3-deficient animals showed an increased survival rate. Additional knock-out of Nox3 in TLR4-deficient animals (TLR4/Nox3 double-deficient mice) nearly rescued the animals comparable to WT controls. Hyperoxia conditions increased macrophage, lymphocyte and neutrophil infiltration into the lungs of WT animals, which was further enhanced in TLR4-deficient animals. Nox3-deficient animals, however, showed no differences compared to WT animals. Notably, TLR4/Nox3 double-deficient animals showed a partial rescue from this phenotype. In WT mice, increased lactate dehydrogenase release as well as increased H2O2 and lipid peroxidation levels were detected in lungs after hyperoxia exposure. TLR4 deficiency further increased these parameters, while Nox3-defcient animals showed reduced levels in comparison to WT animals. These data nicely show that TLR4 signaling somehow inhibits Nox3-mediated ROS production in lungs, which is uncoupled when TLR4 as regulating factor is missing. The Nox3-mediated uncontrolled ROS production then leads to lung destruction. When Nox3, as an ROS source, is removed, it either protects the mice in general from lung injury during hyperoxia, or it leaves the TLR4-dependent inhibition as the terminal factor without any effect. The group also discovered that the Heat Shock Protein 70 (Hsp70) [284][285] is necessary for the TLR4-mediated Nox3 inhibition, since mice and endothelial lung cells deficient for Hsp70 showed increased Nox3 mRNA and protein levels. Notably, mitochondrial matrix O2− levels were decreased in TLR4-deficient lung cells and were not altered in Nox3-deficient cells, excluding mitochondria as a potential ROS source in this setting. In addition, this study firstly investigated possible transcription factors that might influence Nox3 mRNA expression. Chromatin immune-precipitation assays identified regions between −2534/−2360 and −1792/−1498 base pairs upstream of the Nox3 promoter as critical binding sites for STAT3 during Nox3 inhibition. In lungs and endothelial lung cells from endothelial STAT3-deficient mice, more Nox3 expression during both basal and hyperoxia conditions was detected. Electrophoretic Mobility Shift Assay (EMSA) analysis showed that Hsp70 induced the STAT3 binding to the Nox3 promoter region only in WT or Myd88-deficient endothelial lung cells, but not in TLR4- or TRIF-deficient cells. Taken together, this study by Zhang and colleagues is probably the most detailed report about Nox3 activation, regulation and function in a specific context so far. The results were solidified by genetic models and ROS measurements not only in vitro, but also in vivo and no cell type or tissue switching during the study was performed. This is a remarkable example of how to perform a scientific analysis about a Nox enzyme and its functions (Figure 51A,B).
Ruwanpura and colleagues further investigated the role of TLR4 and its adaptors MyD88 adapter-like/Toll/interleukin-1 receptor domain-containing adaptor protein (MAL/TIRAP) [286][287] for normal lung architecture and function in mice [288]. They confirmed the findings from Zhang and colleagues [64], i.e., enlargement of the distal air spaces and destruction of normal alveolar architecture without any inflammation in 6-month-old TLR4-deficient mice. Functionally, they found that the static compliance (pulmonary compliance during the inspiratory pause) was significantly increased in TLR4-deficient mice, which was determined by forced oscillatory technique [289][290][291]. The group further described increased oxidative distress in lung tissue, increased Nox3 mRNA and increased apoptosis of alveolar septal cells. Notably, TLR2 deficiency did not alter any of the observed parameters suggesting a TLR4-specific mechanism in this context.
Yasuoka et al. focused on the influence of ROS during the development of lung fibrosis [292]. During lung diseases, fibrosis is a common side effect, which poses a significant increase in morbidity and mortality in patients [293][294][295]. ROS have been implicated as drivers of fibrosis-related pathophysiology [296][297][298] and lung dysfunction [299][300][301]. Fibrosis is accompanied with tissue remodeling and tissue growth as well as development and is regulated by a plethora of growth factors. Yasukoa et al. focused on the insulin-like growth factor binding protein-5 (IGFBP-5), a prominent factor in this context [302][303], and its connection to lung fibrosis and ROS production. They found that primary human lung fibroblasts increased Nox3 mRNA levels and total cellular ROS production after IGFBP-5 or TGF-β treatment. siRNA-mediated knock-down of Nox3 reduced the ROS production in these cells to baseline levels. However, a role for Nox3-derived ROS in the investigated in vivo setting was not conducted.
The discovery of Nox3 as important player for the progression of lung diseases was furthermore confirmed by a series of genetic screens, which delivered correlative data between the NOX3 gene and different lung diseases. Tremblay et al. conducted a GWAS to identify candidate genes as predisposing factors for genetic asthma association studies [304]. The scan, in combination with the Genes-to-Diseases computational analysis tool [305][306], analyzed 609 subjects from the Saguenay-Lac-St-Jean founder population in Quebec, Canada [307][308]. Amongst several other genes, the NOX3 gene was identified as the only NADPH oxidase-related gene. Yin et al. investigated genetic etiology in the context of non-idiopathic pulmonary hypertension (PH) [309][310][311]. Overall, 208 patients were included, 109 patients were diagnosed with non-idiopathic PH and 99 healthy volunteers were included as controls. A total of 143 SNPs were detected in the 109 PH patients with the top hits located in the chromosome 6, precisely in the locus of the NOX3 gene (SNP termed rs6557421). Notably, PH patients with the detected SNP rs6557421 genotype had a 10-fold-higher risk to develop PH in comparison to healthy control samples. Cantu et al. searched for genetic variations that might increase the risk of primary graft dysfunction (PGD) after lung transplantation by a SNP set analysis [312][313]. Rejection of the grafted lung and subsequent organ dysfunction is a major cause of death during the early transplantational period, affecting up to 30% of all patients [314][315][316]. One of the major pathophysiological aspects associated with PGD is increased oxidative distress occurring during ischemia/reperfusion events [317][318][319][320]. In total, 1039 lung transplant recipients and 392 donors were included in this study, and 314 of the 1038 recipients developed PDG and four genes were identified encoding glutathione peroxidase 1 (GPX1), nuclear factor (erythroid-derived 2)-like 2 (NFE2L2), nitric oxide synthase 3 (NOS3) and glutathione S-transferase mu 2 (GSTM2), which all are involved in antioxidant responses [321][322][323]. In the donor group, the genes for Nox3 (NOX3), nitric oxide synthase 1 adaptor protein (NOS1AP) and paraoxonase 1 (PON1) were associated with the development of PGD. Within the NOX3 gene, the SNP rs3749930 had the strongest association with PGD. The detected SNP marks a nucleotide conversion, which resulted in a threonine to lysine aa substitution in a trans-membrane portion of the Nox3 protein. In addition, several intronic SNPs within the NOX3 gene were associated with increased risk of PGD.
All of these studies clearly demonstrate a critical involvement for Nox3-mediated ROS production as rather destructive factor during lung diseases.
4. Role of Nox3 during Cardiovasclar Diseases
The term “cardiovascular diseases“ summarizes a broad catalogue of diseases that affect one or many components of the cardiovascular system directly. This includes the heart or the blood circulation system, but also simply all other organs and parts of the body as well, since oxygen and nutrient supply, mediated by the blood stream, are crucial for proper functioning of the organism. Thus, this topic intervenes with many other diseases, which are affected by the cardiovascular system. Similar to nearly any other disease outcome, as well as during any kind of cardiovascular disease, increased ROS production is a major contributing factor that worsens diseases progression [324][325][326][327][328][329]. Of course, the involvement of Nox enzymes as ROS sources was intensively investigated, including Nox3 [330].
4.1. Nox3 and Type 2 Diabetes
While fatty acids are crucial components of cellular membranes, chronically increased levels of FFA, consumed with a HFD (Section 5.1.4) lead to obesity due to excessive depositing in non-adipose tissues, e.g. the liver [331][332][333][334]. Subsequently, the development of insulin resistance [335][336], type 2 diabetes [337] and other hepatic diseases [338] dramatically increases. Diabetes mellitus affects more than 300 million people worldwide and represents a disease with high morbidity [339][340]. Type 2 diabetes is associated with various chronic and acute toxic side effects, leading to dyslipidemia, hyperglycemia [341][342][343], diabetic retinopathy [344][345] and chronic hyperinsulinemia. All of these conditions can further induce or enhance adipositas, which is closely related to insulin resistance [346][347][348][349]. Type 2 diabetes and insulin resistance often correlate with increased oxidative distress and an increased systemic pro-inflammatory profile [350] in the according tissues and cells, especially in the liver. Of course, the roles of the Nox isoforms, as primary ROS producers, were investigated in this context [330]. Since Nox3 was identified as an important ROS source in association with diabetic diseases in vitro for HepG2 cells [351][352] (Section 5.1.4) and in a mouse model in vivo [335], further research mostly focused on treatment options. Cremonini et al. investigated the role of the flavanol (-)-Epicatechin [353][354] during HFD-induced insulin-resistance in mice [355]. The group detected a strong up-regulation of Nox3 (60%), Nox4 (274%) and p22phox (237%) protein levels in the liver of mice, which received a HFD in comparison to normally fed mice. Supplemental Epicatechin in the diet prevented this up-regulation. On the in vitro level, similar results were observed in HepG2 cells treated with palmitate and Epicatechin, with exception of p22phox, which remained unaltered. The increased expression of Nox3 and Nox4 resulted in an increased total cellular ROS production. No genetic evidence was provided, and only inhibitors for Nox enzymes were used. Therefore, the specific role of Nox3 or Nox4 could not be determined.
Gupta et al. investigated the effects of Pancreastatin (PST) on adipocyte cells in vitro and in vivo [356]. PST is a peptide secreted by neuroendocrine cells [357], which exploits diabetogenic effects, such as glucose uptake inhibition in liver cells [358][359] or the pancreatic β cell response to insulin [360][361]. Accordingly, treatment with PST is associated with insulin resistance, type 2 diabetes and adipositas [362][363][364]. Since increased ROS levels are involved in lipolysis of adipocytes [246][365] and often correlate with type 2 diabetes progression in patients [330][366][367], the effects of PST on the oxidative distress and chronic insulin induced lipogenesis were also investigated in this study. Neither insulin treatment nor PST treatment alone were sufficient for induction of total cellular ROS production in the adipocyte-like cancer cell line 3T3-L1. Combined treatment induced a slight increase of ROS levels. This corresponded with increased Nox3 protein expression and JNK1/2 phosphorylation. An increase of Nox3 protein expression and JNK1/2 phosphorylation was also detected in white adipose tissue of mice with artificially induced insulin-resistance [368]. While these results nicely contributed to previous findings [335][351][352], no siRNA-mediated knock-down of Nox3 or Nox3-deficient animals were used to clearly confirm Nox3 as the responsible ROS source. Building up from their previous study, the group around Gupta and colleagues researched on possible treatment options with the Pancreastatin inhibitor PSTi8 against insulin resistance [369]. Palmitate treatment of HepG2 cells resulted in lipid accumulation, increased Nox3 mRNA expression, total cellular ROS production and decreased glycogen synthesis. All of these effects were reversed by additional treatment of PSTi. PA also induced phosphorylation of JNK1/2 and p38, which was again prevented by PSTi8 treatment. These findings mark PSTi8 as a potential candidate for diabetic treatment. However, since in both studies, Nox3 was not confirmed as a responsible ROS source, especially since Nox4 is also a prominent ROS source in adipocytes [370][371][372], a clear involvement for Nox3-derived ROS remains elusive.
Malik et al. investigated a previously described therapeutic role of Pterostilbene against insulin resistance [373]. Several studies already described anti-cancer and anti-oxidant effects of Pterostilbene [374], which is a methoxylated Reservatrol analogue [375]. An anti-diabetic effect was also described [376][377][378]. A mechanism of action was not investigated yet. Malik et al. treated HepG2 cells with palmitate, which induced cell death, lipid accumulation, Nox3 mRNA expression, total cellular ROS production and lipid oxidation. Additionally, PA treatment increased expression of genes for proteins involved in fatty acid metabolism, i.e., Sterol regulatory element–binding protein (SREBP1c), Carnitine palmitoyl transferase1 (CPT1), a mitochondrial PA transporter and its transcription factor Peroxisome proliferator-activated receptor alpha (PPARα). All of these effects were strongly reduced after additional treatment with Pterostilbene. While anti-oxidant effects were previously described for Pterostilbene, contradictory, the group observed down-regulation of anti-oxidative enzymes after additional Pterostilbene treatment, therefore outruling an anti-oxidative effect in this context. Since no siRNA knock-down of Nox3 was performed a direct effect of Pterostilbene on Nox3-derived ROS production was not investigated.
Type 2 diabetes negatively affects the outcome of wound healing [379][380] and increased ROS levels correlate with chronic open wounds in patients suffering from Diabetes mellitus [381]. Kim et al. investigated a possible treatment option for improved wound healing [382] by testing the anti-oxidative substance Edaravone. Edaravone was already in use for treatment of acute cerebrovascular diseases [383]. The group used primary human dermal fibroblasts from patients or healthy controls and used the human keratinocyte cell line HaCaT. Furthermore, they conducted a murine in vivo wound healing experiment [384]. Using this model, the group could analyze the expression of Nox3 in tissue flaps near the wound healing area and observed no differences between normo- and hyperglycemic mice after 5 days of operative wound creation. The addition of fibrin for wound healing stimulation or the application of Edaravone did not change Nox3 protein expression. Since no ROS measurements with siRNA knock-down of Nox3 or Nox3 deficient cells were performed, the role of Nox3-derived ROS during the wound healing process remains elusive.
As in the case of lung diseases [304][309][312], also for cardiovascular diseases, GWAS studies were conducted to identify possible risk factors which might influence the disease outcome [385][386][387]. Radowski et al. performed a GWAS to identify genes related to hypertension in 340 patients with type 2 diabetes [388]. Among the six identified genes, the NOX3 gene was also detected, which was previously associated with hypertension [389]. Kwak et al. conducted a GWAS of people with type 2 diabetes to broaden the spectrum of factors, which could help identifying risk factors for cardiovascular diseases in general and type 2 diabetes in particular before the disease outbreak occurs [390]. In their pre-print, they described three variants in genetic loci associated with cardiovascular diseases, especially with type 2 diabetes. Among them, on chromosome 6, there was an intergenic variant between the genes TFB1N and NOX3 (SNP termed rs335407).
4.2. Nox3 and Adipositas
Similar to type 2 diabetes and insulin resistance, ROS also play a role during the inflammatory settings associated with adipositas [346][391][392][393][394]. In adipocytes, the presence of Nox3 was reported before [356]. Issa et al. investigated the influence of cytokines on ROS production and lipolysis in the adipocyte-like cell line 3T3-L1 [246]. Treatment with various pro-inflammatory cytokines (TNF, IL-1β, IFN-γ) induced a slight increase in cellular O2− production after 8 h in differentiated 3T3-L1 cells. It was previously shown that Nox4-derived ROS play an important role for adipocyte differentiation [370][371][372]. Undifferentiated and differentiated 3T3-L1 cells expressed Nox3 as well as Nox4 mRNA. However, only differentiated cells contained the produced Nox3 and Nox4 proteins. While Nox4 expression remained unaltered after cytokine treatment, Nox3 protein levels strongly increased after 8 h. This study nicely showed a decrease in ROS production via Nox3-knockdown after cytokine treatment. Nox-derived ROS were associated with lipolysis in adipocytes before [365] and, indeed, Nox3 knock-down led to an increased lipolysis in 3T3-L1 cells. On the mechanistic level, the group identified an increased phosphorylation of the hormone-sensitive lipase, an enzyme which mediates lipolysis in adipocytes, at the serine residue 536.
4.3. Nox3 and Stroke
Stroke is a major consequence of hypertension [395][396], and elevated ROS levels have been associated with cerebral hemorrhage [397][398][399]. Michihara and colleagues therefore investigated the role of Nox enzymes during stroke development [400]. The group analyzed the cerebrum in a spontaneously hypertensive rat (stroke-prone) model (SHRSP) [401]. These SHRSP animals show lower serum cholesterol levels [402] and increased levels of oxidized proteins in the aorta, heart, kidney [403] and brain [404]. Furthermore, increased 8-OHdG levels in the urine and increased ROS levels in the brain of 16-week-old SHRSP animals were reported [405]. Increased O2− levels, enhanced general Nox activity and increased SOD protein levels were also detected in the brains of SHRSP animals [406]. Michihara et al. analyzed the mRNA levels of Nox enzymes in the cerebrum of SHRSP animals and found increased mRNA levels of Nox2 and Nox3, while Nox1 and Nox4 were not altered and Nox5 was not detected. Notably, Nox3 protein levels were also increased, while Nox2 levels did not change in comparison to the control animals. This is a nice example that both mRNA and protein levels should always be investigated when suggesting changes in protein presence. However, again, no siRNA-mediated knock-down or Nox3 knock-out model was used to provide evidence that Nox3 is the responsible ROS source for the observed effects in SHRSP animals.
4.4. Nox3 and Heart Failure
Several studies have investigated Nox enzymes and their roles for the cardiovascular system in general [407][408][409] and during human [410][411][412][413], mouse [414][415][416][417] and rat heart failure in particular [418][419]. While Nox1, Nox2, Nox4 and Nox5 were detected and investigated in this context, the role of Nox3 remained elusive until its detection in murine embryonic stem cell-derived cardiomyocytes by Li and colleagues [414]. The group mainly detected Nox4 mRNA expression, while Nox3 was only weakly expressed and accordingly focused on Nox4. Bkaily and colleagues further analyzed the role of Nox3 in this setting [420]. For this purpose, they used the hereditary cardiomyopathy hamster model [421][422][423], which is well established for cardiovascular disease studies [424]. They detected Nox1, Nox2 and Nox4, but no Nox3 protein in the ventricular heart muscles of normal hamsters. In the ventricular heart muscles of cardiomyopathic hamsters, they observed a reduction of Nox1 and Nox4 protein levels and an increase of Nox3 protein, while Nox2 levels remained unchanged. These findings nicely demonstrate that Nox isoforms can show a dynamic expression in dependency of the tissue status. The fluctuation of Nox enzyme expression also demonstrates again that siRNA-mediated knock-down or knock-out experiments are strictly needed when claiming a specific role for a certain Nox enzyme as ROS source. Unfortunately, this was also not conducted in this study.
ROS production is also associated with the pathogenesis of ischemia/reperfusion (I/R)-induced heart injuries [425][426] occurring during a myocardial infarction. These injuries include myocardial cell damage and death, arrhythmias or microvascular dysfunction [427][428][429]. Morimoto et al. investigated a putative interplay of ROS and the chemokine monocyte chemoattractant protein-1 (MCP-1) [430][431][432] during I/R [433]. In vitro experiments with neonatal cardiomyocytes showed that under normoxic conditions MCP-1 had no protective effect. However, after I/R induction, apoptotic cell death increased after 6 h and was reduced by treatment with MCP-1. They used Langendorff-perfused mouse hearts from MHC/MCP-1 mice, which overexpress MCP-1 in the heart for further in vivo investigations [434]. The group reported an increase of MCP-1 mRNA and ROS production in WT mice after I/R, which was abolished in hearts from MHC/MCP-1 mice. Notably, the group observed mRNA expression of Nox1, Nox2 and Nox3 in the hearts of WT mice, which decreased after I/R. In the hearts of MHC/MCP-1 mice mRNA levels were lower at basal conditions and rose after I/R, again suggesting a dynamic interplay of Nox-derived ROS production. Unfortunately, no Nox silencing or knock-out neither in vitro nor in vivo was performed. Furthermore, no protein expression was analyzed for Nox3. Hence, if and how Nox3-derived ROS production is activated and if these ROS are involved in the context of MCP-1-mediated cardioprotection could not be clarified.
Vats et al. performed a retrospective cohort study [435] from a population-based Malmö Diet and Cancer Study [436] with 30,446 subjects over 24.3 years. The group analyzed SNPs to detect genetic variations in genes related to oxidative distress and vitamin intake. The study focused on abdominal aortic aneurysm (AAA) [437] and unpredictable ruptured AAA [438][439], both manifesting in an irreversible and life-threatening dilation of the abdominal aorta [437][440][441]. Accordingly, the study only included participants with occurrence of AAA (25,252 patients in total) [436]. Oxidative distress has been suggested as a possible link between various factors that contribute to AAA, such as chronic inflammation and cell death [442][443], with Nox enzymes as correlated endogenous ROS sources [442][444]. During this study, 399 (1.6%) participants were diagnosed with AAA, and 71 (0.2%) were diagnosed with rAAA in general. Furthermore, an amazing effort was made in terms of sub-analytic parameter analysis such as sex, smoking status and physical activity by integrating patient information [445]. The genetic loci were identified by GWAS and altered SNPs for the NOX5 gene (rs150003957), and the NOX3 gene (rs3749930) were detected. The according male patients showed elevated hazard ratios for AAA, while female patients showed no alterations. Furthermore, participants with the dominant NOX3 gene SNP rs3749930 showed an increased risk for rAAA in the overall study. The group additionally performed subgroup analysis to investigate if the detected oxidative distress-related genotypes had an influence on the effect of antioxidant vitamin intake. They reported that men with the NOX3 gene variant rs3749930 showed an inverse association between higher riboflavin vitamin uptake and a hazard risk for intact AAA, which was also confirmed for the overall study population after sex covariate adjusting.
5. Role of Nox3 during Renal Diseases
Chen et al. conducted a GWAS for three phenotypes associated with risk of nephropathy, i.e., serum creatinine levels, creatinine clearance and the glomerular infiltration rate [446][447][448] in 691 type 2 diabetes patients from West Africa to analyze potential factors for reduced renal functions as major consequence of diabetic diseases [389]. The screen detected linkage regions that contain genes, which might influence these three phenotypes. The most prominent candidate genes in these regions that have been implicated in diabetes-induced nephropathy and renal damage were the genes encoding p22phox, (linker region 16q24), Nox1 (linker region 10q22) and Nox3 (linker region 6q25.1–6q26). Together with the study from Ye et al. [449], only two studies investigated Nox3 during kidney-related diseases.
6. Role of Nox3 during Gastrointetinal Disaeses
The most dominant Nox isoform in the gastrointestinal tract is Nox1, which was long termed the “colon NADPH oxidase” [51][450][451]. Nox1 was also detected in the stomach under normal and disease conditions [452][453][454][455]. In addition to Nox1, Duox2 is expressed in the rectum, cecum and ascending colon [456][457][458], and Nox2 and Nox5 were detected in human gastric samples [454]. However, so far, Nox3 has not been detected nor associated with the gastrointestinal tract.
7. Role of Nox3 in Other Diseases
Plantinga et al. investigated genetic variants associated with susceptibility to agranulocytosis [459]. Agranulocytosis is defined as a reduced concentration of granulocytes in peripheral blood (<500 granulocytes/mL blood) [460][461]. Agranulocytosis can be induced by various factors, such as anti-psychotic drugs [462] or antibiotics [463][464], but is also observed in rare events (0.1–0.35%) in patients during treatment with thionamides to medicate hyperthyroidism [465][466][467]. This anti-thyroid drug-induced agranulocytosis (ATDAC) can be a life-threatening condition [461][468], especially after the usage of higher doses of anti-thyroid drugs [469]. During the conducted GWAS of Plantinga et al., two independent families and six patients with Graves’ disease (GD) that developed ATDAC during treatment were analyzed. In 7 out of 11 GD-positive ATDAC patients, a variant of the NOX3 gene were identified. The group reported that the NOX3 gene variants p.Asn8Ser, p.Ala198Thr and p.Arg100Ile were absent in ATDAC-negative GD patients and were not detected in previous genetic screens for predisposition to GD [470][471]. Notably, all variants were located in regions of the membrane-spanning α-helices of the Nox3 protein.
CO poisoning is a consequence of malfunctioning oxygen supply due to carboxyhemoglobin forming in red blood cells [472][473]. The subsequent hypoxia leads to damage in various brain regions, such as the hippocampus or the striatum [474]. However, several research groups have suggested that hypoxia alone cannot be addressed as solely responsible for the brain damage. The involvement of various ROS subspecies has been discussed by Hara et al. and others as possible damage-inducing molecules in this context [475][476][477][478][479]. A previous study already detected increased Duox2 mRNA after CO exposure (3000 ppm, 40 min) in the rat striatum [480], but no mRNA of other Nox isoforms was detected. Hara et al. revised their findings [481] and used their well-established rat model in which CO exposition (1000 ppm or 3000 ppm) [476][482][483] simulates CO poisoning and brain damage [477][484]. The group found a small increase in Nox3 mRNA, while Nox1, Nox2 and Nox4 remained unchanged.
Mikkola et al. performed a GWAS for identification of new gene loci associated with canine hip dysplasia [485]. This canine skeletal disease is a hereditary disorder [486][487] of which the severity varies based on genetic variations [488][489][490] and the dog breed [489][491][492]. The group analyzed 750 German shepherd dogs and identified three new genetic loci associated with this disease. One of these newly identified loci is located on chromosome 1 in an intergenic position between the NOX3 gene and the ARDI1B gene. The group identified the SNP BICF2P468585, which showed the strongest association with the disease and which was located approximately 196 kilobases upstream from the NOX3 gene. Another detected SNP, BICF2S23248027 (also termed rs21911799), was located in the intron between the exons 9 and 10 of the NOX3 gene.
During a study which investigated the therapeutic effects of Dimethyl fumarate (DMF) on relapsing-remitting multiple sclerosis (RRMS) in 564 participants, Carlströem et al. detected a SNP in the NOX3 gene associated with a better DMF treatment outcome [493]. RRMS is an autoimmune disease characterized by the entry of immune cells into the central nervous system (CNS), which leads to pro-inflammatory tissue damage accompanied by neurological dysfunction [494][495]. Like in many other autoimmune pathological settings [496][497], oxidative distress was reported to be a modulating factor in RRMS [498][499][500]. DMF (Tecfidera®) is one of the most prescribed substances for patients that suffer from RRMS [494][501]. The identified SNP rs6919626 in the NOX3 gene allele was associated with a probability of an insufficient DMF treatment response. The group stimulated CD14+ monocytes isolated from patients with the identified NOX3 SNP rs6919626 with Escherichia coli in vitro and detected a reduced total cellular ROS production. This study suggested for the first time a possible link between Nox3-derived ROS and MS disease outcome and treatment.
Li et al. analyzed thyroid tissue samples from 11 patients who suffered from tertiary hyperparathyroidism (THPT) [502]. Hyperparathyroidism manifests itself by an enlargement of the parathyroid gland, increased levels of circulating parathyroid hormone, as well as disturbed bone and mineral metabolism [503][504]. THPT develops during chronic kidney diseases and differs from hyperparathyroidism in an uncontrolled hypercalcemia, i.e., excessive Ca2+ levels in the blood [505]. Since the molecular mechanisms of this process remain largely unknown, Li and colleagues investigated this topic by analyzing blood and thyroid tissue samples from 16 Chinese THPT patients. The group used whole-exome sequencing for the detection of SNPs and insertions or deletions variants. During the screen, 17,401 mutations (6690 missense variants, 3078 frameshift variants, 2005 stop-gained variants and 1630 synonymous variants) were detected in THPT patient samples. From this data set, a further driver mutation analysis identified 179 mutated genes, one of them being the NOX3 gene. Expression quantification by qRT-PCR additionally revealed decreased levels of NOX3 gene mRNA in thyroid gland samples from THPT patients.
References
- Thorne, P.R.; Nuttall, A.L.; Scheibe, F.; Miller, J.M. Sound-induced artifact in cochlear blood flow measurements using the laser doppler flowmeter. Hear. Res. 1987, 31, 229–234.
- Muller, U.; Barr-Gillespie, P.G. New treatment options for hearing loss. Nat. Rev. Drug Discov. 2015, 14, 346–365.
- Petit, C.; Bonnet, C.; Safieddine, S. Deafness: From genetic architecture to gene therapy. Nat. Rev. Genet. 2023, 24, 665–686.
- Kamogashira, T.; Fujimoto, C.; Yamasoba, T. Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. BioMed Res. Int. 2015, 2015, 617207.
- Pittman, C.A.; Ward, B.K.; Nieman, C.L. A review of adult-onset hearing loss: A primer for neurologists. Curr. Treat Options Neurol. 2021, 23, 20.
- Le, T.N.; Straatman, L.V.; Lea, J.; Westerberg, B. Current insights in noise-induced hearing loss: A literature review of the underlying mechanism, pathophysiology, asymmetry, and management options. J. Otolaryngol. Head Neck Surg. Le J. D’oto-Rhino-Laryngol. Chir. Cervico-Faciale 2017, 46, 41.
- Wang, J.; Puel, J.L. Presbycusis: An update on cochlear mechanisms and therapies. J. Clin. Med. 2020, 9, 218.
- Rousset, F.; Nacher-Soler, G.; Coelho, M.; Ilmjarv, S.; Kokje, V.B.C.; Marteyn, A.; Cambet, Y.; Perny, M.; Roccio, M.; Jaquet, V.; et al. Redox activation of excitatory pathways in auditory neurons as mechanism of age-related hearing loss. Redox Biol. 2020, 30, 101434.
- Liberman, M.C.; Kujawa, S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 2017, 349, 138–147.
- Wu, P.Z.; Liberman, L.D.; Bennett, K.; de Gruttola, V.; O’Malley, J.T.; Liberman, M.C. Primary neural degeneration in the human cochlea: Evidence for hidden hearing loss in the aging ear. Neuroscience 2019, 407, 8–20.
- Peineau, T.; Belleudy, S.; Pietropaolo, S.; Bouleau, Y.; Dulon, D. Synaptic release potentiation at aging auditory ribbon synapses. Front. Aging Neurosci. 2021, 13, 756449.
- Frolenkov, G.I.; Belyantseva, I.A.; Friedman, T.B.; Griffith, A.J. Genetic insights into the morphogenesis of inner ear hair cells. Nat. Rev. Genet. 2004, 5, 489–498.
- Schwander, M.; Kachar, B.; Muller, U. Review series: The cell biology of hearing. J. Cell Biol. 2010, 190, 9–20.
- Atkinson, P.J.; Huarcaya Najarro, E.; Sayyid, Z.N.; Cheng, A.G. Sensory hair cell development and regeneration: Similarities and differences. Development 2015, 142, 1561–1571.
- Furness, D.N. Molecular basis of hair cell loss. Cell Tissue Res. 2015, 361, 387–399.
- Kujawa, S.G.; Liberman, M.C. Translating animal models to human therapeutics in noise-induced and age-related hearing loss. Hear. Res. 2019, 377, 44–52.
- Henderson, D.; Bielefeld, E.C.; Harris, K.C.; Hu, B.H. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006, 27, 1–19.
- Ramkumar, V.; Mukherjea, D.; Dhukhwa, A.; Rybak, L.P. Oxidative stress and inflammation caused by cisplatin ototoxicity. Antioxidants 2021, 10, 1919.
- Rousset, F.; Carnesecchi, S.; Senn, P.; Krause, K.H. Nox3-targeted therapies for inner ear pathologies. Curr. Pharm. Des. 2015, 21, 5977–5987.
- Clerici, W.J.; Yang, L. Direct effects of intraperilymphatic reactive oxygen species generation on cochlear function. Hear. Res. 1996, 101, 14–22.
- Bielefeld, E.C.; Hu, B.H.; Harris, K.C.; Henderson, D. Damage and threshold shift resulting from cochlear exposure to paraquat-generated superoxide. Hear. Res. 2005, 207, 35–42.
- Wong, A.C.; Ryan, A.F. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 2015, 7, 58.
- Fetoni, A.R.; Paciello, F.; Rolesi, R.; Paludetti, G.; Troiani, D. Targeting dysregulation of redox homeostasis in noise-induced hearing loss: Oxidative stress and ros signaling. Free Radic. Biol. Med. 2019, 135, 46–59.
- Fujioka, M.; Kanzaki, S.; Okano, H.J.; Masuda, M.; Ogawa, K.; Okano, H. Proinflammatory cytokines expression in noise-induced damaged cochlea. J. Neurosci. Res. 2006, 83, 575–583.
- Kaygusuz, I.; Ozturk, A.; Ustundag, B.; Yalcin, S. Role of free oxygen radicals in noise-related hearing impairment. Hear. Res. 2001, 162, 43–47.
- Yamane, H.; Nakai, Y.; Takayama, M.; Iguchi, H.; Nakagawa, T.; Kojima, A. Appearance of free radicals in the guinea pig inner ear after noise-induced acoustic trauma. Eur. Arch. Oto-Rhino-Laryngol. 1995, 252, 504–508.
- Ohlemiller, K.K.; Wright, J.S.; Dugan, L.L. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol. Neuro-Otol. 1999, 4, 229–236.
- Clerici, W.J.; Hensley, K.; DiMartino, D.L.; Butterfield, D.A. Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear. Res. 1996, 98, 116–124.
- Priuska, E.M.; Schacht, J. Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem. Pharmacol. 1995, 50, 1749–1752.
- Hirose, K.; Hockenbery, D.M.; Rubel, E.W. Reactive oxygen species in chick hair cells after gentamicin exposure in vitro. Hear. Res. 1997, 104, 1–14.
- Dehne, N.; Lautermann, J.; Petrat, F.; Rauen, U.; de Groot, H. Cisplatin ototoxicity: Involvement of iron and enhanced formation of superoxide anion radicals. Toxicol. Appl. Pharmacol. 2001, 174, 27–34.
- Paparella, M.M.; Oda, M.; Hiraide, F.; Brady, D. Pathology of sensorineural hearing loss in otitis media. Ann. Otol. Rhinol. Laryngol. 1972, 81, 632–647.
- Merchant, S.N.; Gopen, Q. A human temporal bone study of acute bacterial meningogenic labyrinthitis. Am. J. Otol. 1996, 17, 375–385.
- Fujimoto, C.; Yamasoba, T. Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxidative Med. Cell. Longev. 2014, 2014, 582849.
- Warraich, U.E.; Hussain, F.; Kayani, H.U.R. Aging—Oxidative stress, antioxidants and computational modeling. Heliyon 2020, 6, e04107.
- Kozakiewicz, M.; Kornatowski, M.; Krzywinska, O.; Kedziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Interv. Aging 2019, 14, 763–771.
- Yamashita, D.; Jiang, H.Y.; Schacht, J.; Miller, J.M. Delayed production of free radicals following noise exposure. Brain Res. 2004, 1019, 201–209.
- Ohlemiller, K.K.; McFadden, S.L.; Ding, D.L.; Lear, P.M.; Ho, Y.S. Targeted mutation of the gene for cellular glutathione peroxidase (gpx1) increases noise-induced hearing loss in mice. J. Assoc. Res. Otolaryngol. JARO 2000, 1, 243–254.
- Yamane, H.; Nakai, Y.; Takayama, M.; Konishi, K.; Iguchi, H.; Nakagawa, T.; Shibata, S.; Kato, A.; Sunami, K.; Kawakatsu, C. The emergence of free radicals after acoustic trauma and strial blood flow. Acta Oto-Laryngol. Suppl. 1995, 519, 87–92.
- Henderson, D.; McFadden, S.L.; Liu, C.C.; Hight, N.; Zheng, X.Y. The role of antioxidants in protection from impulse noise. Ann. N. Y. Acad. Sci. 1999, 884, 368–380.
- Hu, B.H.; Henderson, D.; Nicotera, T.M. Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear. Res. 2002, 166, 62–71.
- Bohne, B. Mechanisms of Noise Damage in the Inner Ear; John H. Mills Raven Press: New York, NY, USA, 1976; pp. 41–68.
- Bohne, B.A.; Harding, G.W.; Lee, S.C. Death pathways in noise-damaged outer hair cells. Hear. Res. 2007, 223, 61–70.
- Murai, N.; Kirkegaard, M.; Jarlebark, L.; Risling, M.; Suneson, A.; Ulfendahl, M. Activation of jnk in the inner ear following impulse noise exposure. J. Neurotrauma 2008, 25, 72–77.
- Seidman, M.D.; Shivapuja, B.G.; Quirk, W.S. The protective effects of allopurinol and superoxide dismutase on noise-induced cochlear damage. Otolaryngol. Head Neck Surg. 1993, 109, 1052–1056.
- Liu, Z. Experimental study on the mechanism of free radical in blast trauma induced hearing loss. Zhonghua Er Bi Yan Hou Ke Za Zhi 1992, 27, 24–26.
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513.
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480.
- Myers, A.L.; Harris, C.M.; Choe, K.M.; Brennan, C.A. Inflammatory production of reactive oxygen species by drosophila hemocytes activates cellular immune defenses. Biochem. Biophys. Res. Commun. 2018, 505, 726–732.
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145.
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226.
- Vermot, A.; Petit-Hartlein, I.; Smith, S.M.E.; Fieschi, F. NADPH oxidases (nox): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890.
- Costa, R.A.; Romagna, C.D.; Pereira, J.L.; Souza-Pinto, N.C. The role of mitochondrial DNA damage in the citotoxicity of reactive oxygen species. J. Bioenerg. Biomembr. 2011, 43, 25–29.
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149.
- Li, P.; Li, S.; Wang, L.; Li, H.; Wang, Y.; Liu, H.; Wang, X.; Zhu, X.; Liu, Z.; Ye, F.; et al. Mitochondrial dysfunction in hearing loss: Oxidative stress, autophagy and nlrp3 inflammasome. Front. Cell Dev. Biol. 2023, 11, 1119773.
- Pak, J.H.; Kim, Y.; Yi, J.; Chung, J.W. Antioxidant therapy against oxidative damage of the inner ear: Protection and preconditioning. Antioxidants 2020, 9, 1076.
- Kishimoto-Urata, M.; Urata, S.; Fujimoto, C.; Yamasoba, T. Role of oxidative stress and antioxidants in acquired inner ear disorders. Antioxidants 2022, 11, 1469.
- Fay, R.R. Comparative psychoacoustics. Hear. Res. 1988, 34, 295–305.
- Mohri, H.; Ninoyu, Y.; Sakaguchi, H.; Hirano, S.; Saito, N.; Ueyama, T. Nox3-derived superoxide in cochleae induces sensorineural hearing loss. J. Neurosci. 2021, 41, 4716–4731.
- Nacher-Soler, G.; Lenglet, S.; Coelho, M.; Thomas, A.; Voruz, F.; Krause, K.H.; Senn, P.; Rousset, F. Local cisplatin delivery in mouse reliably models sensorineural ototoxicity without systemic adverse effects. Front. Cell. Neurosci. 2021, 15, 701783.
- Mukherjea, D.; Jajoo, S.; Kaur, T.; Sheehan, K.E.; Ramkumar, V.; Rybak, L.P. Transtympanic administration of short interfering (si)rna for the nox3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxid. Redox Signal. 2010, 13, 589–598.
- Rousset, F.; Nacher-Soler, G.; Kokje, V.B.C.; Sgroi, S.; Coelho, M.; Krause, K.H.; Senn, P. NADPH oxidase 3 deficiency protects from noise-induced sensorineural hearing loss. Front. Cell Dev. Biol. 2022, 10, 832314.
- Zhang, Y.; Shan, P.; Srivastava, A.; Jiang, G.; Zhang, X.; Lee, P.J. An endothelial hsp70-tlr4 axis limits Nox3 expression and protects against oxidant injury in lungs. Antioxid. Redox Signal. 2016, 24, 991–1012.
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059.
- Nagamani, S.C.; Erez, A.; Eng, C.; Ou, Z.; Chinault, C.; Workman, L.; Coldwell, J.; Stankiewicz, P.; Patel, A.; Lupski, J.R.; et al. Interstitial deletion of 6q25.2-q25.3: A novel microdeletion syndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur. J. Hum. Genet. EJHG 2009, 17, 573–581.
- Milosevic, J.; Kalicanin, P. Long arm deletion of chromosome no. 6 in a mentally retarded boy with multiple physical malformations. J. Ment. Defic. Res. 1975, 19, 139–144.
- Hopkin, R.J.; Schorry, E.; Bofinger, M.; Milatovich, A.; Stern, H.J.; Jayne, C.; Saal, H.M. New insights into the phenotypes of 6q deletions. Am. J. Med. Genet. 1997, 70, 377–386.
- Sukumar, S.; Wang, S.; Hoang, K.; Vanchiere, C.M.; England, K.; Fick, R.; Pagon, B.; Reddy, K.S. Subtle overlapping deletions in the terminal region of chromosome 6q24.2–q26: Three cases studied using fish. Am. J. Med. Genet. 1999, 87, 17–22.
- Pandya, A.; Braverman, N.; Pyeritz, R.E.; Ying, K.L.; Kline, A.D.; Falk, R.E. Interstitial deletion of the long arm of chromosome 6 associated with unusual limb anomalies: Report of two new patients and review of the literature. Am. J. Med. Genet. 1995, 59, 38–43.
- Schuster, M.; Lohscheller, J.; Kummer, P.; Eysholdt, U.; Rosanowski, F. Severe sensory hearing loss in del(6q)-syndrome. Int. J. Pediatr. Otorhinolaryngol. 2003, 67, 1263–1266.
- Stucken, E.Z.; Hong, R.S. Noise-induced hearing loss: An occupational medicine perspective. Curr. Opin. Otolaryngol. Head Neck Surg. 2014, 22, 388–393.
- Dobie, R.A. The burdens of age-related and occupational noise-induced hearing loss in the united states. Ear Hear. 2008, 29, 565–577.
- Yankaskas, K. Prelude: Noise-induced tinnitus and hearing loss in the military. Hear. Res. 2013, 295, 3–8.
- Guski, R.; Schreckenberg, D.; Schuemer, R. Who environmental noise guidelines for the european region: A systematic review on environmental noise and annoyance. Int. J. Environ. Res. Public Health 2017, 14, 1539.
- Saffree Jeffree, M.; Ismail, N.; Awang Lukman, K. Hearing impairment and contributing factors among fertilizer factory workers. J. Occup. Health 2016, 58, 434–443.
- Habybabady, R.H.; Mohammadi, M.; Mortazavi, S.B.; Khavanin, A.; Mirzaei, R.; Malvajerdi, M.S. The effect of simultaneous exposure to cigarette smoke and noise on distortion product otoacoustic emissions in rats. Toxicol. Ind. Health 2019, 35, 349–357.
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 14077–14085.
- Kaur, T.; Clayman, A.C.; Nash, A.J.; Schrader, A.D.; Warchol, M.E.; Ohlemiller, K.K. Lack of fractalkine receptor on macrophages impairs spontaneous recovery of ribbon synapses after moderate noise trauma in c57bl/6 mice. Front. Neurosci. 2019, 13, 620.
- Pourbakht, A.; Yamasoba, T. Cochlear damage caused by continuous and intermittent noise exposure. Hear. Res. 2003, 178, 70–78.
- Op de Beeck, K.; Schacht, J.; Van Camp, G. Apoptosis in acquired and genetic hearing impairment: The programmed death of the hair cell. Hear. Res. 2011, 281, 18–27.
- Frederiksen, T.W.; Ramlau-Hansen, C.H.; Stokholm, Z.A.; Grynderup, M.B.; Hansen, A.M.; Kristiansen, J.; Vestergaard, J.M.; Bonde, J.P.; Kolstad, H.A. Noise-induced hearing loss—A preventable disease? Results of a 10-year longitudinal study of workers exposed to occupational noise. Noise Health 2017, 19, 103–111.
- Shargorodsky, J.; Curhan, S.G.; Curhan, G.C.; Eavey, R. Change in prevalence of hearing loss in us adolescents. JAMA 2010, 304, 772–778.
- Rybak, L.P.; Ramkumar, V. Ototoxicity. Kidney Int. 2007, 72, 931–935.
- Wang, X.; Xu, P.; Li, P.; Wang, Z.; Zhao, F.; Gao, Z.; Xu, L.; Luo, Y.J.; Fan, J.; Liu, P. Alterations in gray matter volume due to unilateral hearing loss. Sci. Rep. 2016, 6, 25811.
- Nakajima, K.; Kanda, E.; Hosobuchi, A.; Suwa, K. Subclinical hearing loss, longer sleep duration, and cardiometabolic risk factors in japanese general population. Int. J. Otolaryngol. 2014, 2014, 218218.
- Lie, A.; Skogstad, M.; Johannessen, H.A.; Tynes, T.; Mehlum, I.S.; Nordby, K.C.; Engdahl, B.; Tambs, K. Occupational noise exposure and hearing: A systematic review. Int. Arch. Occup. Environ. Health 2016, 89, 351–372.
- Sliwinska-Kowalska, M.; Pawelczyk, M. Contribution of genetic factors to noise-induced hearing loss: A human studies review. Mutat. Res. 2013, 752, 61–65.
- Heinonen-Guzejev, M.; Vuorinen, H.S.; Mussalo-Rauhamaa, H.; Heikkilä, K.; Koskenvuo, M.; Kaprio, J. Genetic component of noise sensitivity. Twin Res. Hum. Genet. 2012, 8, 245–249.
- Erway, L.C.; Shiau, Y.W.; Davis, R.R.; Krieg, E.F. Genetics of age-related hearing loss in mice. Iii. Susceptibility of inbred and f1 hybrid strains to noise-induced hearing loss. Hear. Res. 1996, 93, 181–187.
- Kozel, P.J.; Davis, R.R.; Krieg, E.F.; Shull, G.E.; Erway, L.C. Deficiency in plasma membrane calcium atpase isoform 2 increases susceptibility to noise-induced hearing loss in mice. Hear. Res. 2002, 164, 231–239.
- Holme, R.H.; Steel, K.P. Progressive hearing loss and increased susceptibility to noise-induced hearing loss in mice carrying a cdh23 but not a myo7a mutation. J. Assoc. Res. Otolaryngol. JARO 2004, 5, 66–79.
- Ramkumar, V.; Whitworth, C.A.; Pingle, S.C.; Hughes, L.F.; Rybak, L.P. Noise induces a1 adenosine receptor expression in the chinchilla cochlea. Hear. Res. 2004, 188, 47–56.
- Yang, W.; Vethanayagam, R.R.; Dong, Y.; Cai, Q.; Hu, B.H. Activation of the antigen presentation function of mononuclear phagocyte populations associated with the basilar membrane of the cochlea after acoustic overstimulation. Neuroscience 2015, 303, 1–15.
- Hashimoto, S.; Billings, P.; Harris, J.P.; Firestein, G.S.; Keithley, E.M. Innate immunity contributes to cochlear adaptive immune responses. Audiol. Neuro-Otol. 2005, 10, 35–43.
- Wakabayashi, K.; Fujioka, M.; Kanzaki, S.; Okano, H.J.; Shibata, S.; Yamashita, D.; Masuda, M.; Mihara, M.; Ohsugi, Y.; Ogawa, K.; et al. Blockade of interleukin-6 signaling suppressed cochlear inflammatory response and improved hearing impairment in noise-damaged mice cochlea. Neurosci. Res. 2010, 66, 345–352.
- Keithley, E.M.; Wang, X.; Barkdull, G.C. Tumor necrosis factor alpha can induce recruitment of inflammatory cells to the cochlea. Otol. Neurotol. 2008, 29, 854–859.
- Paciello, F.; Di Pino, A.; Rolesi, R.; Troiani, D.; Paludetti, G.; Grassi, C.; Fetoni, A.R. Anti-oxidant and anti-inflammatory effects of caffeic acid: In Vivo evidences in a model of noise-induced hearing loss. Food Chem. Toxicol. 2020, 143, 111555.
- Kurabi, A.; Keithley, E.M.; Housley, G.D.; Ryan, A.F.; Wong, A.C. Cellular mechanisms of noise-induced hearing loss. Hear. Res. 2017, 349, 129–137.
- Bennett, B.J.; Farber, C.R.; Orozco, L.; Kang, H.M.; Ghazalpour, A.; Siemers, N.; Neubauer, M.; Neuhaus, I.; Yordanova, R.; Guan, B.; et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010, 20, 281–290.
- Farber, C.R.; Bennett, B.J.; Orozco, L.; Zou, W.; Lira, A.; Kostem, E.; Kang, H.M.; Furlotte, N.; Berberyan, A.; Ghazalpour, A.; et al. Mouse genome-wide association and systems genetics identify asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 2011, 7, e1002038.
- Park, C.C.; Gale, G.D.; de Jong, S.; Ghazalpour, A.; Bennett, B.J.; Farber, C.R.; Langfelder, P.; Lin, A.; Khan, A.H.; Eskin, E.; et al. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst. Biol. 2011, 5, 43.
- Lavinsky, J.; Crow, A.L.; Pan, C.; Wang, J.; Aaron, K.A.; Ho, M.K.; Li, Q.; Salehide, P.; Myint, A.; Monges-Hernadez, M.; et al. Genome-wide association study identifies Nox3 as a critical gene for susceptibility to noise-induced hearing loss. PLoS Genet. 2015, 11, e1005094.
- Zhao, T.; Wang, Y.; Li, Z.; Xu, X.; Lei, S.; Huang, L.; Xu, L.; Zhang, M.; Yang, L. Associations of noise kurtosis, genetic variations in nox3 and lifestyle factors with noise-induced hearing loss. Environ. Health A Glob. Access Sci. Source 2020, 19, 13.
- Lei, S.F.; Ahroon, W.A.; Hamernik, R.P. The application of frequency and time domain kurtosis to the assessment of hazardous noise exposures. J. Acoust. Soc. Am. 1994, 96, 1435–1444.
- Zhao, Y.M.; Qiu, W.; Zeng, L.; Chen, S.S.; Cheng, X.R.; Davis, R.I.; Hamernik, R.P. Application of the kurtosis statistic to the evaluation of the risk of hearing loss in workers exposed to high-level complex noise. Ear Hear. 2010, 31, 527–532.
- Sato, S.; Kitamura, T.; Sakai, H.; Ando, Y. The loudness of “complex noise” in relation to the factors extracted from the auto-correlation function. J. Sound Vib. 2001, 241, 97–103.
- Erdreich, J. A distribution based definition of impulse noise. J. Acoust. Soc. Am. 1986, 79, 990–998.
- Xie, H.W.; Qiu, W.; Heyer, N.J.; Zhang, M.B.; Zhang, P.; Zhao, Y.M.; Hamernik, R.P. The use of the kurtosis-adjusted cumulative noise exposure metric in evaluating the hearing loss risk for complex noise. Ear Hear. 2016, 37, 312–323.
- Davis, R.I.; Qiu, W.; Heyer, N.J.; Zhao, Y.; Qiuling Yang, M.S.; Li, N.; Tao, L.; Zhu, L.; Zeng, L.; Yao, D. The use of the kurtosis metric in the evaluation of occupational hearing loss in workers in china: Implications for hearing risk assessment. Noise Health 2012, 14, 330–342.
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. Nox3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072.
- Flaherty, J.P.; Fairfield, H.E.; Spruce, C.A.; McCarty, C.M.; Bergstrom, D.E. Molecular characterization of an allelic series of mutations in the mouse Nox3 gene. Mamm. Genome 2011, 22, 156–169.
- Du, Z.; Li, S.; Liu, L.; Yang, Q.; Zhang, H.; Gao, C. NADPH oxidase 3-associated oxidative stress and caspase 3-dependent apoptosis in the cochleae of d-galactose-induced aged rats. Mol. Med. Rep. 2015, 12, 7883–7890.
- Vlajkovic, S.M.; Lin, S.C.; Wong, A.C.; Wackrow, B.; Thorne, P.R. Noise-induced changes in expression levels of NADPH oxidases in the cochlea. Hear. Res. 2013, 304, 145–152.
- Dhukhwa, A.; Bhatta, P.; Sheth, S.; Korrapati, K.; Tieu, C.; Mamillapalli, C.; Ramkumar, V.; Mukherjea, D. Targeting inflammatory processes mediated by trpvi and tnf-alpha for treating noise-induced hearing loss. Front. Cell. Neurosci. 2019, 13, 444.
- Kaur, T.; Borse, V.; Sheth, S.; Sheehan, K.; Ghosh, S.; Tupal, S.; Jajoo, S.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Adenosine a1 receptor protects against cisplatin ototoxicity by suppressing the nox3/stat1 inflammatory pathway in the cochlea. J. Neurosci. 2016, 36, 3962–3977.
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem. J. 2007, 403, 97–108.
- Paffenholz, R.; Bergstrom, R.A.; Pasutto, F.; Wabnitz, P.; Munroe, R.J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J.; et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004, 18, 486–491.
- Kiss, P.J.; Knisz, J.; Zhang, Y.; Baltrusaitis, J.; Sigmund, C.D.; Thalmann, R.; Smith, R.J.; Verpy, E.; Banfi, B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr. Biol. CB 2006, 16, 208–213.
- Zahra, G.; Esmaeil, K.; Mohammad, F.; Rashidy-Pour, A.; Mahdi, M.; Mahdi, A.; Ali, K. Combined effects of the exposure to silver nanoparticles and noise on hearing function and cochlea structure of the male rats. Life Sci. 2022, 304, 120724.
- Peralta-Videa, J.R.; Zhao, L.; Lopez-Moreno, M.L.; de la Rosa, G.; Hong, J.; Gardea-Torresdey, J.L. Nanomaterials and the environment: A review for the biennium 2008–2010. J. Hazard. Mater. 2011, 186, 1–15.
- Roda, E.; Bottone, M.G.; Biggiogera, M.; Milanesi, G.; Coccini, T. Pulmonary and hepatic effects after low dose exposure to nanosilver: Early and long-lasting histological and ultrastructural alterations in rat. Toxicol. Rep. 2019, 6, 1047–1060.
- Recordati, C.; De Maglie, M.; Bianchessi, S.; Argentiere, S.; Cella, C.; Mattiello, S.; Cubadda, F.; Aureli, F.; D’Amato, M.; Raggi, A. Tissue distribution and acute toxicity of silver after single intravenous administration in mice: Nano-specific and size-dependent effects. Part. Fibre Toxicol. 2015, 13, 12.
- Xu, L.; Li, X.; Takemura, T.; Hanagata, N.; Wu, G.; Chou, L.L. Genotoxicity and molecular response of silver nanoparticle (np)-based hydrogel. J. Nanobiotechnol. 2012, 10, 16.
- Ferdous, Z.; Nemmar, A. Health impact of silver nanoparticles: A review of the biodistribution and toxicity following various routes of exposure. Int. J. Mol. Sci. 2020, 21, 2375.
- AshaRani, P.V.; Low Kah Mun, G.; Hande, M.P.; Valiyaveettil, S. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano 2009, 3, 279–290.
- Kawata, K.; Osawa, M.; Okabe, S. In vitro toxicity of silver nanoparticles at noncytotoxic doses to hepg2 human hepatoma cells. Environ. Sci. Technol. 2009, 43, 6046–6051.
- Kim, H.R.; Kim, M.J.; Lee, S.Y.; Oh, S.M.; Chung, K.H. Genotoxic effects of silver nanoparticles stimulated by oxidative stress in human normal bronchial epithelial (beas-2b) cells. Mutat. Res. 2011, 726, 129–135.
- Neely, S.T.; Johnson, T.A.; Kopun, J.; Dierking, D.M.; Gorga, M.P. Distortion-product otoacoustic emission input/output characteristics in normal-hearing and hearing-impaired human ears. J. Acoust. Soc. Am. 2009, 126, 728–738.
- Martin, G.K.; Stagner, B.B.; Chung, Y.S.; Lonsbury-Martin, B.L. Characterizing distortion-product otoacoustic emission components across four species. J. Acoust. Soc. Am. 2011, 129, 3090–3103.
- Shahtaheri, S.J.; Goodarzi, Z.; Karami, E.; Khavanin, A.; Khansari, M.G.; Kiani, M.; Rashidy-Pour, A. Effects of acute exposure to al(2)o(3)-nps (alpha and gamma) and white noise and their combination on cochlea structure and function in wistar rats. Environ. Sci. Pollut. Res. Int. 2023, 30, 89859–89876.
- Ates, M.; Demir, V.; Arslan, Z.; Daniels, J.; Farah, I.O.; Bogatu, C. Evaluation of alpha and gamma aluminum oxide nanoparticle accumulation, toxicity, and depuration in artemia salina larvae. Environ. Toxicol. 2015, 30, 109–118.
- Patlolla, A.K.; Kumari, S.A.; Madhusudhanachary, P.; Turner, T.; Tchounwou, P.B. Biochemical and histopathological evaluation of al(2)o(3) nanomaterials in kidney of wistar rats. Curr. Top. Biochem. Res. 2018, 19, 1–12.
- Jacukowicz-Sobala, I.; Ocinski, D.; Kociolek-Balawejder, E. Iron and aluminium oxides containing industrial wastes as adsorbents of heavy metals: Application possibilities and limitations. Waste Manag. Res. 2015, 33, 612–629.
- Brown, D.M.; Brown, A.M.; Willitsford, A.H.; Dinello-Fass, R.; Airola, M.B.; Siegrist, K.M.; Thomas, M.E.; Chang, Y. Lidar measurements of solid rocket propellant fire particle plumes. Appl. Opt. 2016, 55, 4657–4669.
- Hunter, D.; Milton, R.; Perry, K.M.A.; Thompson, D.R. Effect of aluminium and alumina on the lung in grinders of duralumin aeroplane propellers. Br. J. Ind. Med. 1944, 1, 159–164.
- Sikkeland, L.; Alexis, N.E.; Fry, R.C.; Martin, E.; Danielsen, T.E.; Sostrand, P.; Kongerud, J. Inflammation in induced sputum after aluminium oxide exposure: An experimental chamber study. Occup. Environ. Med. 2016, 73, 199–205.
- Xing, M.; Zou, H.; Gao, X.; Chang, B.; Tang, S.; Zhang, M. Workplace exposure to airborne alumina nanoparticles associated with separation and packaging processes in a pilot factory. Environ. Sci. Process Impacts 2015, 17, 656–666.
- Lu, J.; Li, W.; Du, X.; Ewert, D.L.; West, M.B.; Stewart, C.; Floyd, R.A.; Kopke, R.D. Antioxidants reduce cellular and functional changes induced by intense noise in the inner ear and cochlear nucleus. J. Assoc. Res. Otolaryngol. JARO 2014, 15, 353–372.
- Fram, R.J. Cisplatin and platinum analogues: Recent advances. Curr. Opin. Oncol. 1992, 4, 1073–1079.
- Boulikas, T.; Vougiouka, M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review). Oncol. Rep. 2004, 11, 559–595.
- Laurell, G. Pharmacological intervention in the field of ototoxicity. Hno 2019, 67, 434–439.
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320.
- Jordan, J.A.; Schwade, N.D.; Truelson, J.M. Fosfomycin does not inhibit the tumoricidal efficacy of cisplatinum. Laryngoscope 1999, 109, 1259–1262.
- Saleh, S.; El-Demerdash, E. Protective effects of l-arginine against cisplatin-induced renal oxidative stress and toxicity: Role of nitric oxide. Basic Clin. Pharmacol. Toxicol. 2005, 97, 91–97.
- Bodenner, D.L.; Dedon, P.C.; Keng, P.C.; Borch, R.F. Effect of diethyldithiocarbamate on cis-diamminedichloroplatinum(ii)-induced cytotoxicity, DNA cross-linking, and gamma-glutamyl transpeptidase inhibition. Cancer Res. 1986, 46, 2745–2750.
- Barabas, K.; Milner, R.; Lurie, D.; Adin, C. Cisplatin: A review of toxicities and therapeutic applications. Vet. Comp. Oncol. 2008, 6, 1–18.
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901.
- Schacht, J.; Talaska, A.E.; Rybak, L.P. Cisplatin and aminoglycoside antibiotics: Hearing loss and its prevention. Anat. Rec. 2012, 295, 1837–1850.
- Koike, N.; Sasaki, A.; Murakami, T.; Suzuki, K. Effect of edaravone against cisplatin-induced chronic renal injury. Drug Chem. Toxicol. 2021, 44, 437–446.
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460.
- Rybak, L.P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Cisplatin ototoxicity and protection: Clinical and experimental studies. Tohoku J. Exp. Med. 2009, 219, 177–186.
- McKeage, M.J. Comparative adverse effect profiles of platinum drugs. Drug Saf. 1995, 13, 228–244.
- Bokemeyer, C.; Berger, C.C.; Hartmann, J.T.; Kollmannsberger, C.; Schmoll, H.J.; Kuczyk, M.A.; Kanz, L. Analysis of risk factors for cisplatin-induced ototoxicity in patients with testicular cancer. Br. J. Cancer 1998, 77, 1355–1362.
- Huang, E.; Teh, B.S.; Strother, D.R.; Davis, Q.G.; Chiu, J.K.; Lu, H.H.; Carpenter, L.S.; Mai, W.Y.; Chintagumpala, M.M.; South, M.; et al. Intensity-modulated radiation therapy for pediatric medulloblastoma: Early report on the reduction of ototoxicity. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 599–605.
- Anniko, M.; Sobin, A. Cisplatin: Evaluation of its ototoxic potential. Am. J. Otolaryngol. 1986, 7, 276–293.
- Tsukasaki, N.; Whitworth, C.A.; Rybak, L.P. Acute changes in cochlear potentials due to cisplatin. Hear. Res. 2000, 149, 189–198.
- Rybak, L.P.; Kelly, T. Ototoxicity: Bioprotective mechanisms. Curr. Opin. Otolaryngol. Head Neck Surg. 2003, 11, 328–333.
- Watanabe, K.I.; Hess, A.; Bloch, W.; Michel, O. Nitric oxide synthase inhibitor suppresses the ototoxic side effect of cisplatin in guinea pigs. Anticancer Drugs 2000, 11, 401–406.
- Hong, O.; Kerr, M.J.; Poling, G.L.; Dhar, S. Understanding and preventing noise-induced hearing loss. Dis.-A-Mon. DM 2013, 59, 110–118.
- Kopke, R.D.; Liu, W.; Gabaizadeh, R.; Jacono, A.; Feghali, J.; Spray, D.; Garcia, P.; Steinman, H.; Malgrange, B.; Ruben, R.J.; et al. Use of organotypic cultures of corti’s organ to study the protective effects of antioxidant molecules on cisplatin-induced damage of auditory hair cells. Am. J. Otol. 1997, 18, 559–571.
- Rybak, L.P.; Husain, K.; Whitworth, C.; Somani, S.M. Dose dependent protection by lipoic acid against cisplatin-induced ototoxicity in rats: Antioxidant defense system. Toxicol. Sci. Off. J. Soc. Toxicol. 1999, 47, 195–202.
- Waissbluth, S.; Salehi, P.; He, X.; Daniel, S.J. Systemic dexamethasone for the prevention of cisplatin-induced ototoxicity. Eur. Arch. Oto-Rhino-Laryngol. 2013, 270, 1597–1605.
- Rybak, L.P. Cis-platinum associated hearing loss. J. Laryngol. Otol. 1981, 95, 745–747.
- Fleischman, R.W.; Stadnicki, S.W.; Ethier, M.F.; Schaeppi, U. Ototoxicity of cis-dichlorodiammine platinum (ii) in the guinea pig. Toxicol. Appl. Pharmacol. 1975, 33, 320–332.
- Stadnicki, S.W.; Fleischman, R.W.; Schaeppi, U.; Merriam, P. Cis-dichlorodiammineplatinum (ii) (nsc-119875): Hearing loss and other toxic effects in rhesus monkeys. Cancer Chemother. Rep. 1975, 59, 467–480.
- Staecker, H.; Liu, W.; Malgrange, B.; Lefebvre, P.P.; Van De Water, T.R. Vector-mediated delivery of bcl-2 prevents degeneration of auditory hair cells and neurons after injury. ORL 2007, 69, 43–50.
- Zhang, N.; Cai, J.; Xu, L.; Wang, H.; Liu, W. Cisplatin-induced stria vascularis damage is associated with inflammation and fibrosis. Neural Plast. 2020, 2020, 8851525.
- Sluyter, S.; Klis, S.F.; de Groot, J.C.; Smoorenburg, G.F. Alterations in the stria vascularis in relation to cisplatin ototoxicity and recovery. Hear. Res. 2003, 185, 49–56.
- Kanzaki, J.; Ouchi, T. Steroid-responsive bilateral sensorineural hearing loss and immune complexes. Arch. Oto-Rhino-Laryngol. 1981, 230, 5–9.
- Yoshida, K.; Ichimiya, I.; Suzuki, M.; Mogi, G. Effect of proinflammatory cytokines on cultured spiral ligament fibrocytes. Hear. Res. 1999, 137, 155–159.
- Satoh, H.; Firestein, G.S.; Billings, P.B.; Harris, J.P.; Keithley, E.M. Proinflammatory cytokine expression in the endolymphatic sac during inner ear inflammation. J. Assoc. Res. Otolaryngol. JARO 2003, 4, 139–147.
- Kim, H.J.; So, H.S.; Lee, J.H.; Park, C.; Lee, J.B.; Youn, M.J.; Kim, S.J.; Yang, S.H.; Lee, K.M.; Kwon, K.B.; et al. Role of proinflammatory cytokines in cisplatin-induced vestibular hair cell damage. Head Neck 2008, 30, 1445–1456.
- So, H.; Kim, H.; Kim, Y.; Kim, E.; Pae, H.O.; Chung, H.T.; Kim, H.J.; Kwon, K.B.; Lee, K.M.; Lee, H.Y.; et al. Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via nrf2/ho-1. J. Assoc. Res. Otolaryngol. JARO 2008, 9, 290–306.
- Watanabe, K.; Inai, S.; Jinnouchi, K.; Bada, S.; Hess, A.; Michel, O.; Yagi, T. Nuclear-factor kappa b (nf-kappa b)-inducible nitric oxide synthase (inos/nos ii) pathway damages the stria vascularis in cisplatin-treated mice. Anticancer. Res. 2002, 22, 4081–4085.
- Ries, F.; Klastersky, J. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am. J. Kidney Dis. 1986, 8, 368–379.
- Leibbrandt, M.E.; Wolfgang, G.H.; Metz, A.L.; Ozobia, A.A.; Haskins, J.R. Critical subcellular targets of cisplatin and related platinum analogs in rat renal proximal tubule cells. Kidney Int. 1995, 48, 761–770.
- Sugiyama, S.; Hayakawa, M.; Kato, T.; Hanaki, Y.; Shimizu, K.; Ozawa, T. Adverse effects of anti-tumor drug, cisplatin, on rat kidney mitochondria: Disturbances in glutathione peroxidase activity. Biochem. Biophys. Res. Commun. 1989, 159, 1121–1127.
- So, H.S.; Park, C.; Kim, H.J.; Lee, J.H.; Park, S.Y.; Lee, J.H.; Lee, Z.W.; Kim, H.M.; Kalinec, F.; Lim, D.J.; et al. Protective effect of t-type calcium channel blocker flunarizine on cisplatin-induced death of auditory cells. Hear. Res. 2005, 204, 127–139.
- Goncalves, M.S.; Silveira, A.F.; Teixeira, A.R.; Hyppolito, M.A. Mechanisms of cisplatin ototoxicity: Theoretical review. J. Laryngol. Otol. 2013, 127, 536–541.
- Matsushima, H.; Yonemura, K.; Ohishi, K.; Hishida, A. The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J. Lab. Clin. Med. 1998, 131, 518–526.
- Rybak, L.P.; Whitworth, C.; Somani, S. Application of antioxidants and other agents to prevent cisplatin ototoxicity. Laryngoscope 1999, 109, 1740–1744.
- Sergi, B.; Ferraresi, A.; Troiani, D.; Paludetti, G.; Fetoni, A.R. Cisplatin ototoxicity in the guinea pig: Vestibular and cochlear damage. Hear. Res. 2003, 182, 56–64.
- Takumida, M.; Anniko, M. Simultaneous detection of both nitric oxide and reactive oxygen species in guinea pig vestibular sensory cells. ORL 2002, 64, 143–147.
- Darlington, C.L.; Smith, P.F. Vestibulotoxicity following aminoglycoside antibiotics and its prevention. Curr. Opin. Investig. Drugs 2003, 4, 841–846.
- Rivolta, M.N.; Grix, N.; Lawlor, P.; Ashmore, J.F.; Jagger, D.J.; Holley, M.C. Auditory hair cell precursors immortalized from the mammalian inner ear. Proceedings. Biol. Sci. 1998, 265, 1595–1603.
- Mukherjea, D.; Jajoo, S.; Whitworth, C.; Bunch, J.R.; Turner, J.G.; Rybak, L.P.; Ramkumar, V. Short interfering rna against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 13056–13065.
- Kim, H.J.; Lee, J.H.; Kim, S.J.; Oh, G.S.; Moon, H.D.; Kwon, K.B.; Park, C.; Park, B.H.; Lee, H.K.; Chung, S.Y.; et al. Roles of NADPH oxidases in cisplatin-induced reactive oxygen species generation and ototoxicity. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3933–3946.
- Mukherjea, D.; Whitworth, C.A.; Nandish, S.; Dunaway, G.A.; Rybak, L.P.; Ramkumar, V. Expression of the kidney injury molecule 1 in the rat cochlea and induction by cisplatin. Neuroscience 2006, 139, 733–740.
- Benkafadar, N.; Menardo, J.; Bourien, J.; Nouvian, R.; Francois, F.; Decaudin, D.; Maiorano, D.; Puel, J.L.; Wang, J. Reversible p53 inhibition prevents cisplatin ototoxicity without blocking chemotherapeutic efficacy. EMBO Mol. Med. 2017, 9, 7–26.
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International union of basic and clinical pharmacology. Lxxxi. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34.
- Ramkumar, V.; Ravi, R.; Wilson, M.C.; Gettys, T.W.; Whitworth, C.; Rybak, L.P. Identification of a1 adenosine receptors in rat cochlea coupled to inhibition of adenylyl cyclase. Am. J. Physiol. 1994, 267, C731–C737.
- Ford, M.S.; Nie, Z.; Whitworth, C.; Rybak, L.P.; Ramkumar, V. Up-regulation of adenosine receptors in the cochlea by cisplatin. Hear. Res. 1997, 111, 143–152.
- Ford, M.S.; Maggirwar, S.B.; Rybak, L.P.; Whitworth, C.; Ramkumar, V. Expression and function of adenosine receptors in the chinchilla cochlea. Hear. Res. 1997, 105, 130–140.
- Bryant, G.M.; Barron, S.E.; Norris, C.H.; Guth, P.S. Adenosine is a modulator of hair cell-afferent neurotransmission. Hear. Res. 1987, 30, 231–237.
- Nario, K.; Kitano, I.; Mori, N.; Matsunaga, T. The effect of adenosine on cochlear potentials in the guinea pig. Eur. Arch. Oto-Rhino-Laryngol. 1994, 251, 428–433.
- Hight, N.G.; McFadden, S.L.; Henderson, D.; Burkard, R.F.; Nicotera, T. Noise-induced hearing loss in chinchillas pre-treated with glutathione monoethylester and r-pia. Hear. Res. 2003, 179, 21–32.
- Vlajkovic, S.M.; Guo, C.X.; Telang, R.; Wong, A.C.; Paramananthasivam, V.; Boison, D.; Housley, G.D.; Thorne, P.R. Adenosine kinase inhibition in the cochlea delays the onset of age-related hearing loss. Exp. Gerontol. 2011, 46, 905–914.
- Whitworth, C.A.; Ramkumar, V.; Jones, B.; Tsukasaki, N.; Rybak, L.P. Protection against cisplatin ototoxicity by adenosine agonists. Biochem. Pharmacol. 2004, 67, 1801–1807.
- Kaur, T.; Mukherjea, D.; Sheehan, K.; Jajoo, S.; Rybak, L.P.; Ramkumar, V. Short interfering rna against stat1 attenuates cisplatin-induced ototoxicity in the rat by suppressing inflammation. Cell Death Dis. 2011, 2, e180.
- Demmler, G.J. Infectious diseases society of america and centers for disease control. Summary of a workshop on surveillance for congenital cytomegalovirus disease. Rev. Infect. Dis. 1991, 13, 315–329.
- Stagno, S.; Pass, R.F.; Cloud, G.; Britt, W.J.; Henderson, R.E.; Walton, P.D.; Veren, D.A.; Page, F.; Alford, C.A. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 1986, 256, 1904–1908.
- Barbi, M.; Binda, S.; Caroppo, S.; Primache, V. Neonatal screening for congenital cytomegalovirus infection and hearing loss. J. Clin. Virol. 2006, 35, 206–209.
- Dreher, A.M.; Arora, N.; Fowler, K.B.; Novak, Z.; Britt, W.J.; Boppana, S.B.; Ross, S.A. Spectrum of disease and outcome in children with symptomatic congenital cytomegalovirus infection. J. Pediatr. 2014, 164, 855–859.
- Shi, X.; Dong, Y.; Li, Y.; Zhao, Z.; Li, H.; Qiu, S.; Li, Y.; Guo, W.; Qiao, Y. Inflammasome activation in mouse inner ear in response to mcmv induced hearing loss. J. Otol. 2015, 10, 143–149.
- Zhuang, W.; Wang, C.; Shi, X.; Qiu, S.; Zhang, S.; Xu, B.; Chen, M.; Jiang, W.; Dong, H.; Qiao, Y. Mcmv triggers ros/nlrp3-associated inflammasome activation in the inner ear of mice and cultured spiral ganglion neurons, contributing to sensorineural hearing loss. Int. J. Mol. Med. 2018, 41, 3448–3456.
- Schachtele, S.J.; Mutnal, M.B.; Schleiss, M.R.; Lokensgard, J.R. Cytomegalovirus-induced sensorineural hearing loss with persistent cochlear inflammation in neonatal mice. J. Neurovirol. 2011, 17, 201–211.
- Neag, M.A.; Mocan, A.; Echeverria, J.; Pop, R.M.; Bocsan, C.I.; Crisan, G.; Buzoianu, A.D. Berberine: Botanical occurrence, traditional uses, extraction methods, and relevance in cardiovascular, metabolic, hepatic, and renal disorders. Front. Pharmacol. 2018, 9, 557.
- Zhuang, W.; Li, T.; Wang, C.; Shi, X.; Li, Y.; Zhang, S.; Zhao, Z.; Dong, H.; Qiao, Y. Berberine exerts antioxidant effects via protection of spiral ganglion cells against cytomegalovirus-induced apoptosis. Free Radic. Biol. Med. 2018, 121, 127–135.
- Katsarkas, A.; Ayukawa, H. Hearing loss due to aging (presbycusis). J. Otolaryngol. 1986, 15, 239–244.
- Gennis, V.; Garry, P.J.; Haaland, K.Y.; Yeo, R.A.; Goodwin, J.S. Hearing and cognition in the elderly. New findings and a review of the literature. Arch. Intern. Med. 1991, 151, 2259–2264.
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120.
- Panza, F.; Solfrizzi, V.; Logroscino, G. Age-related hearing impairment-a risk factor and frailty marker for dementia and ad. Nat. Rev. Neurol. 2015, 11, 166–175.
- Rutherford, B.R.; Brewster, K.; Golub, J.S.; Kim, A.H.; Roose, S.P. Sensation and psychiatry: Linking age-related hearing loss to late-life depression and cognitive decline. Am. J. Psychiatry 2018, 175, 215–224.
- Yang, C.H.; Schrepfer, T.; Schacht, J. Age-related hearing impairment and the triad of acquired hearing loss. Front. Cell. Neurosci. 2015, 9, 276.
- Scholtz, A.W.; Kammen-Jolly, K.; Felder, E.; Hussl, B.; Rask-Andersen, H.; Schrott-Fischer, A. Selective aspects of human pathology in high-tone hearing loss of the aging inner ear. Hear. Res. 2001, 157, 77–86.
- Chen, B.; Zhong, Y.; Peng, W.; Sun, Y.; Kong, W.J. Age-related changes in the central auditory system: Comparison of d-galactose-induced aging rats and naturally aging rats. Brain Res. 2010, 1344, 43–53.
- Chen, B.; Zhong, Y.; Peng, W.; Sun, Y.; Hu, Y.J.; Yang, Y.; Kong, W.J. Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of d-galactose-induced aging rats. Mol. Biol. Rep. 2011, 38, 3635–3642.
- Du, Z.; Yang, Y.; Hu, Y.; Sun, Y.; Zhang, S.; Peng, W.; Zhong, Y.; Huang, X.; Kong, W. A long-term high-fat diet increases oxidative stress, mitochondrial damage and apoptosis in the inner ear of d-galactose-induced aging rats. Hear. Res. 2012, 287, 15–24.
- Cui, X.; Zuo, P.; Zhang, Q.; Li, X.; Hu, Y.; Long, J.; Packer, L.; Liu, J. Chronic systemic d-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: Protective effects of r-alpha-lipoic acid. J. Neurosci. Res. 2006, 83, 1584–1590.
- Hua, X.; Lei, M.; Zhang, Y.; Ding, J.; Han, Q.; Hu, G.; Xiao, M. Long-term d-galactose injection combined with ovariectomy serves as a new rodent model for alzheimer’s disease. Life Sci. 2007, 80, 1897–1905.
- Lu, J.; Zheng, Y.L.; Wu, D.M.; Luo, L.; Sun, D.X.; Shan, Q. Ursolic acid ameliorates cognition deficits and attenuates oxidative damage in the brain of senescent mice induced by d-galactose. Biochem. Pharmacol. 2007, 74, 1078–1090.
- Chen, C.F.; Lang, S.Y.; Zuo, P.P.; Yang, N.; Wang, X.Q.; Xia, C. Effects of d-galactose on the expression of hippocampal peripheral-type benzodiazepine receptor and spatial memory performances in rats. Psychoneuroendocrinology 2006, 31, 805–811.
- Kumar, A.; Prakash, A.; Dogra, S. Naringin alleviates cognitive impairment, mitochondrial dysfunction and oxidative stress induced by d-galactose in mice. Food Chem. Toxicol. 2010, 48, 626–632.
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F. Purple sweet potato color alleviates d-galactose-induced brain aging in old mice by promoting survival of neurons via pi3k pathway and inhibiting cytochrome c-mediated apoptosis. Brain Pathol. 2010, 20, 598–612.
- Zhang, Z.F.; Lu, J.; Zheng, Y.L.; Hu, B.; Fan, S.H.; Wu, D.M.; Zheng, Z.H.; Shan, Q.; Liu, C.M. Purple sweet potato color protects mouse liver against d-galactose-induced apoptosis via inhibiting caspase-3 activation and enhancing pi3k/akt pathway. Food Chem. Toxicol. 2010, 48, 2500–2507.
- Lei, M.; Hua, X.; Xiao, M.; Ding, J.; Han, Q.; Hu, G. Impairments of astrocytes are involved in the d-galactose-induced brain aging. Biochem. Biophys. Res. Commun. 2008, 369, 1082–1087.
- Hsieh, H.M.; Wu, W.M.; Hu, M.L. Soy isoflavones attenuate oxidative stress and improve parameters related to aging and alzheimer’s disease in c57bl/6j mice treated with d-galactose. Food Chem. Toxicol. 2009, 47, 625–632.
- Cui, X.; Wang, L.; Zuo, P.; Han, Z.; Fang, Z.; Li, W.; Liu, J. D-galactose-caused life shortening in drosophila melanogaster and musca domestica is associated with oxidative stress. Biogerontology 2004, 5, 317–325.
- Kong, W.J.; Wang, Y.; Wang, Q.; Hu, Y.J.; Han, Y.C.; Liu, J. The relation between d-galactose injection and mitochondrial DNA 4834 bp deletion mutation. Exp. Gerontol. 2006, 41, 628–634.
- Kong, W.J.; Hu, Y.J.; Wang, Q.; Wang, Y.; Han, Y.C.; Cheng, H.M.; Kong, W.; Guan, M.X. The effect of the mtdna4834 deletion on hearing. Biochem. Biophys. Res. Commun. 2006, 344, 425–430.
- Zhong, Y.; Hu, Y.J.; Yang, Y.; Peng, W.; Sun, Y.; Chen, B.; Huang, X.; Kong, W.J. Contribution of common deletion to total deletion burden in mitochondrial DNA from inner ear of d-galactose-induced aging rats. Mutat. Res. 2011, 712, 11–19.
- Zhong, Y.; Hu, Y.J.; Chen, B.; Peng, W.; Sun, Y.; Yang, Y.; Zhao, X.Y.; Fan, G.R.; Huang, X.; Kong, W.J. Mitochondrial transcription factor a overexpression and base excision repair deficiency in the inner ear of rats with d-galactose-induced aging. FEBS J. 2011, 278, 2500–2510.
- Yamasoba, T.; Someya, S.; Yamada, C.; Weindruch, R.; Prolla, T.A.; Tanokura, M. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear. Res. 2007, 226, 185–193.
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390.
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652.
- Markaryan, A.; Nelson, E.G.; Hinojosa, R. Quantification of the mitochondrial DNA common deletion in presbycusis. Laryngoscope 2009, 119, 1184–1189.
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33.
- Basu, S.; Xie, X.; Uhler, J.P.; Hedberg-Oldfors, C.; Milenkovic, D.; Baris, O.R.; Kimoloi, S.; Matic, S.; Stewart, J.B.; Larsson, N.G.; et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLoS Genet. 2020, 16, e1009242.
- Schon, E.A.; Rizzuto, R.; Moraes, C.T.; Nakase, H.; Zeviani, M.; DiMauro, S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 1989, 244, 346–349.
- Lee, H.C.; Pang, C.Y.; Hsu, H.S.; Wei, Y.H. Differential accumulations of 4,977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta 1994, 1226, 37–43.
- Mohamed, S.A.; Hanke, T.; Erasmi, A.W.; Bechtel, M.J.; Scharfschwerdt, M.; Meissner, C.; Sievers, H.H.; Gosslau, A. Mitochondrial DNA deletions and the aging heart. Exp. Gerontol. 2006, 41, 508–517.
- Nakano, Y.; Longo-Guess, C.M.; Bergstrom, D.E.; Nauseef, W.M.; Jones, S.M.; Banfi, B. Mutation of the cyba gene encoding p22phox causes vestibular and immune defects in mice. J. Clin. Investig. 2008, 118, 1176–1185.
- Chen, P.; Chai, Y.; Liu, H.; Li, G.; Wang, L.; Yang, T.; Wu, H. Postnatal development of microglia-like cells in mouse cochlea. Neural Plast. 2018, 2018, 1970150.
- Sun, S.; Yu, H.; Yu, H.; Honglin, M.; Ni, W.; Zhang, Y.; Guo, L.; He, Y.; Xue, Z.; Ni, Y.; et al. Inhibition of the activation and recruitment of microglia-like cells protects against neomycin-induced ototoxicity. Mol. Neurobiol. 2015, 51, 252–267.
- Morioka, S.; Sakaguchi, H.; Yamaguchi, T.; Ninoyu, Y.; Mohri, H.; Nakamura, T.; Hisa, Y.; Ogita, K.; Saito, N.; Ueyama, T. Hearing vulnerability after noise exposure in a mouse model of reactive oxygen species overproduction. J. Neurochem. 2018, 146, 459–473.
- Issa, N.; Lachance, G.; Bellmann, K.; Laplante, M.; Stadler, K.; Marette, A. Cytokines promote lipolysis in 3t3-l1 adipocytes through induction of NADPH oxidase 3 expression and superoxide production. J. Lipid Res. 2018, 59, 2321–2328.
- Brault, J.; Vigne, B.; Meunier, M.; Beaumel, S.; Mollin, M.; Park, S.; Stasia, M.J. Nox4 is the main NADPH oxidase involved in the early stages of hematopoietic differentiation from human induced pluripotent stem cells. Free Radic. Biol. Med. 2020, 146, 107–118.
- Chen, G.; Gharib, T.G.; Huang, C.C.; Taylor, J.M.; Misek, D.E.; Kardia, S.L.; Giordano, T.J.; Iannettoni, M.D.; Orringer, M.B.; Hanash, S.M.; et al. Discordant protein and mrna expression in lung adenocarcinomas. Mol. Cell Proteom. 2002, 1, 304–313.
- Pascal, L.E.; True, L.D.; Campbell, D.S.; Deutsch, E.W.; Risk, M.; Coleman, I.M.; Eichner, L.J.; Nelson, P.S.; Liu, A.Y. Correlation of mrna and protein levels: Cell type-specific gene expression of cluster designation antigens in the prostate. BMC Genom. 2008, 9, 246.
- Gluschko, A.; Herb, M.; Wiegmann, K.; Krut, O.; Neiss, W.F.; Utermohlen, O.; Kronke, M.; Schramm, M. The β2 integrin mac-1 induces protective lc3-associated phagocytosis of listeria monocytogenes. Cell Host Microbe 2018, 23, 324–337.e325.
- Rousset, F.; Salmon, P.; Bredl, S.; Cherpin, O.; Coelho, M.; Myburgh, R.; Alessandrini, M.; Perny, M.; Roccio, M.; Speck, R.F.; et al. Optimizing synthetic mirna minigene architecture for efficient mirna hairpin concatenation and multi-target gene knockdown. Mol. Ther. Nucleic Acids 2019, 14, 351–363.
- Zhang, S.; Xing, J.; Gong, Y.; Li, P.; Wang, B.; Xu, L. Downregulation of vdr in benign paroxysmal positional vertigo patients inhibits otolith-associated protein expression levels. Mol. Med. Rep. 2021, 24, 591.
- Froehling, D.A.; Silverstein, M.D.; Mohr, D.N.; Beatty, C.W.; Offord, K.P.; Ballard, D.J. Benign positional vertigo: Incidence and prognosis in a population-based study in olmsted county, minnesota. Mayo Clin. Proc. 1991, 66, 596–601.
- Imai, T.; Higashi-Shingai, K.; Takimoto, Y.; Masumura, C.; Hattori, K.; Inohara, H. New scoring system of an interview for the diagnosis of benign paroxysmal positional vertigo. Acta Oto-Laryngol. 2016, 136, 283–288.
- Fife, T.D.; Iverson, D.J.; Lempert, T.; Furman, J.M.; Baloh, R.W.; Tusa, R.J.; Hain, T.C.; Herdman, S.; Morrow, M.J.; Gronseth, G.S.; et al. Practice parameter: Therapies for benign paroxysmal positional vertigo (an evidence-based review): Report of the quality standards subcommittee of the american academy of neurology. Neurology 2008, 70, 2067–2074.
- Bhattacharyya, N.; Gubbels, S.P.; Schwartz, S.R.; Edlow, J.A.; El-Kashlan, H.; Fife, T.; Holmberg, J.M.; Mahoney, K.; Hollingsworth, D.B.; Roberts, R.; et al. Clinical practice guideline: Benign paroxysmal positional vertigo (update). Otolaryngol.-Head Neck Surg. 2017, 156, S1–S47.
- Lundberg, Y.W.; Xu, Y.; Thiessen, K.D.; Kramer, K.L. Mechanisms of otoconia and otolith development. Dev. Dyn. 2015, 244, 239–253.
- Jang, Y.S.; Hwang, C.H.; Shin, J.Y.; Bae, W.Y.; Kim, L.S. Age-related changes on the morphology of the otoconia. Laryngoscope 2006, 116, 996–1001.
- Baloh, R.W.; Honrubia, V.; Jacobson, K. Benign positional vertigo: Clinical and oculographic features in 240 cases. Neurology 1987, 37, 371–378.
- Nakashima, T.; Pyykko, I.; Arroll, M.A.; Casselbrant, M.L.; Foster, C.A.; Manzoor, N.F.; Megerian, C.A.; Naganawa, S.; Young, Y.H. Meniere’s disease. Nat. Rev. Dis. Primers 2016, 2, 16028.
- Jeong, S.H.; Kim, J.S.; Shin, J.W.; Kim, S.; Lee, H.; Lee, A.Y.; Kim, J.M.; Jo, H.; Song, J.; Ghim, Y. Decreased serum vitamin d in idiopathic benign paroxysmal positional vertigo. J. Neurol. 2013, 260, 832–838.
- Wu, Y.; Gu, C.; Han, W.; Lu, X.; Chen, C.; Fan, Z. Reduction of bone mineral density in native chinese female idiopathic benign paroxysmal positional vertigo patients. Am J Otolaryngol 2018, 39, 31–33.
- Yoda, S.; Cureoglu, S.; Yildirim-Baylan, M.; Morita, N.; Fukushima, H.; Harada, T.; Paparella, M.M. Association between type 1 diabetes mellitus and deposits in the semicircular canals. Otolaryngol. Head Neck Surg. 2011, 145, 458–462.
- Holick, M.F. Resurrection of vitamin d deficiency and rickets. J. Clin. Investig. 2006, 116, 2062–2072.
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin d: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 2016, 96, 365–408.
- Editorial Board of Chinese Journal of Otorhinolaryngology Head and Neck Surgery; Society of Otorhinolaryngology Head and Neck Surgery Chinese Medical Association. Guideline of diagnosis and treatment of benign paroxysmal positional vertigo (2017). Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi Chin. J. Otorhinolaryngol. Head Neck Surg. 2017, 52, 173–177.
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688.
- Lokke, A.; Lange, P.; Scharling, H.; Fabricius, P.; Vestbo, J. Developing copd: A 25 year follow up study of the general population. Thorax 2006, 61, 935–939.
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259.
- MacNee, W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 50–60.
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265.
- Zhang, X.; Shan, P.; Sasidhar, M.; Chupp, G.L.; Flavell, R.A.; Choi, A.M.; Lee, P.J. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 305–315.
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Iizuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975.
- Pons, J.; Sauleda, J.; Regueiro, V.; Santos, C.; Lopez, M.; Ferrer, J.; Agusti, A.G.; Bengoechea, J.A. Expression of toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir. Res. 2006, 7, 64.
- Sukkar, M.B.; Xie, S.; Khorasani, N.M.; Kon, O.M.; Stanbridge, R.; Issa, R.; Chung, K.F. Toll-like receptor 2, 3, and 4 expression and function in human airway smooth muscle. J. Allergy Clin. Immunol. 2006, 118, 641–648.
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249.
- Chung, K.F.; Marwick, J.A. Molecular mechanisms of oxidative stress in airways and lungs with reference to asthma and chronic obstructive pulmonary disease. Ann. N. Y. Acad. Sci. 2010, 1203, 85–91.
- Pratico, D.; Basili, S.; Vieri, M.; Cordova, C.; Violi, F.; Fitzgerald, G.A. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane f2alpha-iii, an index of oxidant stress. Am. J. Respir. Crit. Care Med. 1998, 158, 1709–1714.
- Lius, E.E.; Syafaah, I. Hyperoxia in the management of respiratory failure: A literature review. Ann. Med. Surg. 2022, 81, 104393.
- Lilien, T.A.; van Meenen, D.M.P.; Schultz, M.J.; Bos, L.D.J.; Bem, R.A. Hyperoxia-induced lung injury in acute respiratory distress syndrome: What is its relative impact? Am. J. Physiol. Lung Cell. Mol. Physiol. 2023, 325, L9–L16.
- Minkove, S.; Dhamapurkar, R.; Cui, X.; Li, Y.; Sun, J.; Cooper, D.; Eichacker, P.Q.; Torabi-Parizi, P. Effect of low-to-moderate hyperoxia on lung injury in preclinical animal models: A systematic review and meta-analysis. Intensive Care Med. Exp. 2023, 11, 22.
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141.
- Zhang, X.; Shan, P.; Qureshi, S.; Homer, R.; Medzhitov, R.; Noble, P.W.; Lee, P.J. Cutting edge: Tlr4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 2005, 175, 4834–4838.
- Zhang, Y.; Zhang, X.; Shan, P.; Hunt, C.R.; Pandita, T.K.; Lee, P.J. A protective hsp70-tlr4 pathway in lethal oxidant lung injury. J. Immunol. 2013, 191, 1393–1403.
- Hunt, C.R.; Dix, D.J.; Sharma, G.G.; Pandita, R.K.; Gupta, A.; Funk, M.; Pandita, T.K. Genomic instability and enhanced radiosensitivity in hsp70.1- and hsp70.3-deficient mice. Mol. Cell. Biol. 2004, 24, 899–911.
- Fitzgerald, K.A.; Palsson-McDermott, E.M.; Bowie, A.G.; Jefferies, C.A.; Mansell, A.S.; Brady, G.; Brint, E.; Dunne, A.; Gray, P.; Harte, M.T.; et al. Mal (myd88-adapter-like) is required for toll-like receptor-4 signal transduction. Nature 2001, 413, 78–83.
- Rajpoot, S.; Wary, K.K.; Ibbott, R.; Liu, D.; Saqib, U.; Thurston, T.L.M.; Baig, M.S. Tirap in the mechanism of inflammation. Front. Immunol. 2021, 12, 697588.
- Ruwanpura, S.M.; McLeod, L.; Lilja, A.R.; Brooks, G.; Dousha, L.F.; Seow, H.J.; Bozinovski, S.; Vlahos, R.; Hertzog, P.J.; Anderson, G.P.; et al. Non-essential role for tlr2 and its signaling adaptor mal/tirap in preserving normal lung architecture in mice. PLoS ONE 2013, 8, e78095.
- Ruwanpura, S.M.; McLeod, L.; Miller, A.; Jones, J.; Bozinovski, S.; Vlahos, R.; Ernst, M.; Armes, J.; Bardin, P.G.; Anderson, G.P.; et al. Interleukin-6 promotes pulmonary emphysema associated with apoptosis in mice. Am. J. Respir. Cell Mol. Biol. 2011, 45, 720–730.
- Ruwanpura, S.M.; McLeod, L.; Miller, A.; Jones, J.; Vlahos, R.; Ramm, G.; Longano, A.; Bardin, P.G.; Bozinovski, S.; Anderson, G.P.; et al. Deregulated stat3 signaling dissociates pulmonary inflammation from emphysema in gp130 mutant mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L627–L639.
- Hantos, Z.; Adamicza, A.; Janosi, T.Z.; Szabari, M.V.; Tolnai, J.; Suki, B. Lung volumes and respiratory mechanics in elastase-induced emphysema in mice. J. Appl. Physiol. 2008, 105, 1864–1872.
- Yasuoka, H.; Garrett, S.M.; Nguyen, X.X.; Artlett, C.M.; Feghali-Bostwick, C.A. NADPH oxidase-mediated induction of reactive oxygen species and extracellular matrix deposition by insulin-like growth factor binding protein-5. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L644–L655.
- King, T.E., Jr.; Schwarz, M.I.; Brown, K.; Tooze, J.A.; Colby, T.V.; Waldron, J.A., Jr.; Flint, A.; Thurlbeck, W.; Cherniack, R.M. Idiopathic pulmonary fibrosis: Relationship between histopathologic features and mortality. Am. J. Respir. Crit. Care Med. 2001, 164, 1025–1032.
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011, 66, 462–467.
- Zisman, D.A.; Keane, M.P.; Belperio, J.A.; Strieter, R.M.; Lynch, J.P. Pulmonary fibrosis. In Fibrosis Research: Methods and Protocols; Varga, J., Brenner, D.A., Phan, S.H., Eds.; Humana Press: Totowa, NJ, USA, 2005; pp. 3–44.
- Bocchino, M.; Agnese, S.; Fagone, E.; Svegliati, S.; Grieco, D.; Vancheri, C.; Gabrielli, A.; Sanduzzi, A.; Avvedimento, E.V. Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS ONE 2010, 5, e14003.
- Manoury, B.; Nenan, S.; Leclerc, O.; Guenon, I.; Boichot, E.; Planquois, J.M.; Bertrand, C.P.; Lagente, V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir. Res. 2005, 6, 11.
- Murthy, S.; Adamcakova-Dodd, A.; Perry, S.S.; Tephly, L.A.; Keller, R.M.; Metwali, N.; Meyerholz, D.K.; Wang, Y.; Glogauer, M.; Thorne, P.S.; et al. Modulation of reactive oxygen species by rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L846–L855.
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422.
- Korfei, M.; von der Beck, D.; Henneke, I.; Markart, P.; Ruppert, C.; Mahavadi, P.; Ghanim, B.; Klepetko, W.; Fink, L.; Meiners, S.; et al. Comparative proteome analysis of lung tissue from patients with idiopathic pulmonary fibrosis (ipf), non-specific interstitial pneumonia (nsip) and organ donors. J. Proteom. 2013, 85, 109–128.
- Montuschi, P.; Ciabattoni, G.; Paredi, P.; Pantelidis, P.; du Bois, R.M.; Kharitonov, S.A.; Barnes, P.J. 8-isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1998, 158, 1524–1527.
- Beattie, J.; Allan, G.J.; Lochrie, J.D.; Flint, D.J. Insulin-like growth factor-binding protein-5 (igfbp-5): A critical member of the igf axis. Biochem. J. 2006, 395, 1–19.
- Han, N.; Zhang, F.; Li, G.; Zhang, X.; Lin, X.; Yang, H.; Wang, L.; Cao, Y.; Du, J.; Fan, Z. Local application of igfbp5 protein enhanced periodontal tissue regeneration via increasing the migration, cell proliferation and osteo/dentinogenic differentiation of mesenchymal stem cells in an inflammatory niche. Stem Cell Res. Ther. 2017, 8, 210.
- Tremblay, K.; Lemire, M.; Potvin, C.; Tremblay, A.; Hunninghake, G.M.; Raby, B.A.; Hudson, T.J.; Perez-Iratxeta, C.; Andrade-Navarro, M.A.; Laprise, C. Genes to diseases (g2d) computational method to identify asthma candidate genes. PLoS ONE 2008, 3, e2907.
- Perez-Iratxeta, C.; Wjst, M.; Bork, P.; Andrade, M.A. G2d: A tool for mining genes associated with disease. BMC Genet 2005, 6, 45.
- Perez-Iratxeta, C.; Bork, P.; Andrade-Navarro, M.A. Update of the g2d tool for prioritization of gene candidates to inherited diseases. Nucleic Acids Res. 2007, 35, W212–W216.
- Heyer, E.; Tremblay, M. Variability of the genetic contribution of quebec population founders associated to some deleterious genes. Am. J. Hum. Genet. 1995, 56, 970–978.
- Richter, A.; Rioux, J.D.; Bouchard, J.P.; Mercier, J.; Mathieu, J.; Ge, B.; Poirier, J.; Julien, D.; Gyapay, G.; Weissenbach, J.; et al. Location score and haplotype analyses of the locus for autosomal recessive spastic ataxia of charlevoix-saguenay, in chromosome region 13q11. Am. J. Hum. Genet. 1999, 64, 768–775.
- Yin, C.; Li, K.; Yu, Y.; Huang, H.; Yu, Y.; Wang, Z.; Yan, J.; Pu, Y.; Li, Z.; Li, D.; et al. Genome-wide association study identifies loci and candidate genes for non-idiopathic pulmonary hypertension in eastern chinese han population. BMC Pulm. Med. 2018, 18, 158.
- Barman, S.A.; Chen, F.; Li, X.; Haigh, S.; Stepp, D.W.; Kondrikov, D.; Mahboubi, K.; Bordan, Z.; Traber, P.; Su, Y.; et al. Galectin-3 promotes vascular remodeling and contributes to pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 1488–1492.
- Soubrier, F.; Chung, W.K.; Machado, R.; Grunig, E.; Aldred, M.; Geraci, M.; Loyd, J.E.; Elliott, C.G.; Trembath, R.C.; Newman, J.H.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D13–D21.
- Cantu, E.; Shah, R.J.; Lin, W.; Daye, Z.J.; Diamond, J.M.; Suzuki, Y.; Ellis, J.H.; Borders, C.F.; Andah, G.A.; Beduhn, B.; et al. Oxidant stress regulatory genetic variation in recipients and donors contributes to risk of primary graft dysfunction after lung transplantation. J. Thorac. Cardiovasc. Surg. 2015, 149, 596–602.
- Charles, E.J.; Kron, I.L. One step closer to the elimination of primary graft dysfunction. J. Thorac. Cardiovasc. Surg. 2015, 149, 602–603.
- Christie, J.D.; Carby, M.; Bag, R.; Corris, P.; Hertz, M.; Weill, D. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction Part II: Definition. A consensus statement of the international society for heart and lung transplantation. J. Heart Lung Transplant 2005, 24, 1454–1459.
- Christie, J.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Aurora, P.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Stehlik, J.; Hertz, M.I. The registry of the international society for heart and lung transplantation: Twenty-seventh official adult lung and heart-lung transplant report—2010. J. Heart Lung Transplant. 2010, 29, 1104–1118.
- Suzuki, Y.; Cantu, E.; Christie, J.D. Primary graft dysfunction. Semin. Respir. Crit. Care Med. 2013, 34, 305–319.
- de Perrot, M.; Liu, M.; Waddell, T.K.; Keshavjee, S. Ischemia-reperfusion-induced lung injury. Am. J. Respir. Crit. Care Med. 2003, 167, 490–511.
- Williams, A.; Riise, G.C.; Anderson, B.A.; Kjellstrom, C.; Schersten, H.; Kelly, F.J. Compromised antioxidant status and persistent oxidative stress in lung transplant recipients. Free Radic. Res. 1999, 30, 383–393.
- Rega, F.R.; Wuyts, W.A.; Vanaudenaerde, B.M.; Jannis, N.C.; Neyrinck, A.P.; Verleden, G.M.; Lerut, T.E.; Van Raemdonck, D.E. Nebulized n-acetyl cysteine protects the pulmonary graft inside the non-heart-beating donor. J. Heart Lung Transplant. 2005, 24, 1369–1377.
- Kozower, B.D.; Christofidou-Solomidou, M.; Sweitzer, T.D.; Muro, S.; Buerk, D.G.; Solomides, C.C.; Albelda, S.M.; Patterson, G.A.; Muzykantov, V.R. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nat. Biotechnol. 2003, 21, 392–398.
- Cho, H.Y.; Reddy, S.P.; Kleeberger, S.R. Nrf2 defends the lung from oxidative stress. Antioxid. Redox Signal. 2006, 8, 76–87.
- Lubos, E.; Mahoney, C.E.; Leopold, J.A.; Zhang, Y.Y.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 modulates lipopolysaccharide-induced adhesion molecule expression in endothelial cells by altering cd14 expression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2525–2532.
- Hamanishi, T.; Furuta, H.; Kato, H.; Doi, A.; Tamai, M.; Shimomura, H.; Sakagashira, S.; Nishi, M.; Sasaki, H.; Sanke, T.; et al. Functional variants in the glutathione peroxidase-1 (gpx-1) gene are associated with increased intima-media thickness of carotid arteries and risk of macrovascular diseases in japanese type 2 diabetic patients. Diabetes 2004, 53, 2455–2460.
- Liu, P.; Shi, L.; Cang, X.; Huang, J.; Wu, X.; Yan, J.; Chen, L.; Cui, S.; Ye, X. Ctbp2 ameliorates palmitate-induced insulin resistance in hepg2 cells through ros mediated jnk pathway. Gen. Comp. Endocrinol. 2017, 247, 66–73.
- Styskal, J.; Van Remmen, H.; Richardson, A.; Salmon, A.B. Oxidative stress and diabetes: What can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic. Biol. Med. 2012, 52, 46–58.
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575.
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999.
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metab. Clin. Exp. 2008, 57, 1071–1077.
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; Van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792.
- Elnakish, M.T.; Hassanain, H.H.; Janssen, P.M.; Angelos, M.G.; Khan, M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: Important role of rac/NADPH oxidase. J. Pathol. 2013, 231, 290–300.
- Zhang, C.H.; Zhou, B.G.; Sheng, J.Q.; Chen, Y.; Cao, Y.Q.; Chen, C. Molecular mechanisms of hepatic insulin resistance in nonalcoholic fatty liver disease and potential treatment strategies. Pharmacol. Res. 2020, 159, 104984.
- Santoleri, D.; Titchenell, P.M. Resolving the paradox of hepatic insulin resistance. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 447–456.
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Investig. 2002, 32, 14–23.
- Collaboration, N.C.D.R.F. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396.
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH oxidase 3-derived reactive oxygen species through jnk and p38mapk pathways. J. Biol. Chem. 2010, 285, 29965–29973.
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in h4iiec3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818.
- Montonen, J.; Knekt, P.; Harkanen, T.; Jarvinen, R.; Heliovaara, M.; Aromaa, A.; Reunanen, A. Dietary patterns and the incidence of type 2 diabetes. Am. J. Epidemiol. 2005, 161, 219–227.
- Parks, E.; Yki-Jarvinen, H.; Hawkins, M. Out of the frying pan: Dietary saturated fat influences nonalcoholic fatty liver disease. J. Clin. Investig. 2017, 127, 454–456.
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14.
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the international diabetes federation diabetes atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843.
- Magnusson, I.; Rothman, D.L.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Increased rate of gluconeogenesis in type ii diabetes mellitus. A 13c nuclear magnetic resonance study. J. Clin. Investig. 1992, 90, 1323–1327.
- Bock, G.; Chittilapilly, E.; Basu, R.; Toffolo, G.; Cobelli, C.; Chandramouli, V.; Landau, B.R.; Rizza, R.A. Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose: Role of increased rates of gluconeogenesis. Diabetes 2007, 56, 1703–1711.
- Basu, R.; Barosa, C.; Jones, J.; Dube, S.; Carter, R.; Basu, A.; Rizza, R.A. Pathogenesis of prediabetes: Role of the liver in isolated fasting hyperglycemia and combined fasting and postprandial hyperglycemia. J. Clin. Endocrinol. Metab. 2013, 98, E409–E417.
- Funatsu, H.; Yamashita, H. Molecular biology in development and progression of diabetic retinopathy. Nihon Rinsho Jpn. J. Clin. Med. 2002, 60, 162–166.
- Wilkinson, C.P.; Ferris, F.L., 3rd; Klein, R.E.; Lee, P.P.; Agardh, C.D.; Davis, M.; Dills, D.; Kampik, A.; Pararajasegaram, R.; Verdaguer, J.T.; et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 2003, 110, 1677–1682.
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948.
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for jnk in obesity and insulin resistance. Nature 2002, 420, 333–336.
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857.
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122.
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7.
- Li, L.; He, Q.; Huang, X.; Man, Y.; Zhou, Y.; Wang, S.; Wang, J.; Li, J. Nox3-derived reactive oxygen species promote tnf-alpha-induced reductions in hepatocyte glycogen levels via a JNK pathway. FEBS Lett. 2010, 584, 995–1000.
- Carnesecchi, S.; Carpentier, J.L.; Foti, M.; Szanto, I. Insulin-induced vascular endothelial growth factor expression is mediated by the NADPH oxidase Nox3. Exp. Cell Res. 2006, 312, 3413–3424.
- Bettaieb, A.; Vazquez Prieto, M.A.; Rodriguez Lanzi, C.; Miatello, R.M.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. (-)-epicatechin mitigates high-fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free Radic. Biol. Med. 2014, 72, 247–256.
- Bettaieb, A.; Cremonini, E.; Kang, H.; Kang, J.; Haj, F.G.; Oteiza, P.I. Anti-inflammatory actions of (-)-epicatechin in the adipose tissue of obese mice. Int. J. Biochem. Cell Biol. 2016, 81, 383–392.
- Cremonini, E.; Wang, Z.; Bettaieb, A.; Adamo, A.M.; Daveri, E.; Mills, D.A.; Kalanetra, K.M.; Haj, F.G.; Karakas, S.; Oteiza, P.I. (-)-epicatechin protects the intestinal barrier from high fat diet-induced permeabilization: Implications for steatosis and insulin resistance. Redox Biol. 2018, 14, 588–599.
- Gupta, A.P.; Syed, A.A.; Garg, R.; Goand, U.K.; Singh, P.; Riyazuddin, M.; Valicherla, G.R.; Husain, A.; Gayen, J.R. Pancreastatin inhibitor psti8 attenuates hyperinsulinemia induced obesity and inflammation mediated insulin resistance via mapk/nox3-jnk pathway. Eur. J. Pharmacol. 2019, 864, 172723.
- Loh, Y.P.; Cheng, Y.; Mahata, S.K.; Corti, A.; Tota, B. Chromogranin a and derived peptides in health and disease. J. Mol. Neurosci. MN 2012, 48, 347–356.
- Hossain, Z.; Valicherla, G.R.; Gupta, A.P.; Syed, A.A.; Riyazuddin, M.; Chandra, S.; Siddiqi, M.I.; Gayen, J.R. Discovery of pancreastatin inhibitor psti8 for the treatment of insulin resistance and diabetes: Studies in rodent models of diabetes mellitus. Sci. Rep. 2018, 8, 8715.
- Sanchez-Margalet, V.; Gonzalez-Yanes, C. Pancreastatin inhibits insulin action in rat adipocytes. Am. J. Physiol. 1998, 275, E1055–E1060.
- Gayen, J.R.; Saberi, M.; Schenk, S.; Biswas, N.; Vaingankar, S.M.; Cheung, W.W.; Najjar, S.M.; O’Connor, D.T.; Bandyopadhyay, G.; Mahata, S.K. A novel pathway of insulin sensitivity in chromogranin a null mice: A crucial role for pancreastatin in glucose homeostasis. J. Biol. Chem. 2009, 284, 28498–28509.
- Tatemoto, K.; Efendic, S.; Mutt, V.; Makk, G.; Feistner, G.J.; Barchas, J.D. Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion. Nature 1986, 324, 476–478.
- Bandyopadhyay, G.K.; Lu, M.; Avolio, E.; Siddiqui, J.A.; Gayen, J.R.; Wollam, J.; Vu, C.U.; Chi, N.W.; O’Connor, D.T.; Mahata, S.K. Pancreastatin-dependent inflammatory signaling mediates obesity-induced insulin resistance. Diabetes 2015, 64, 104–116.
- Broedbaek, K.; Hilsted, L. Chromogranin a as biomarker in diabetes. Biomark. Med. 2016, 10, 1181–1189.
- Allu, P.K.; Chirasani, V.R.; Ghosh, D.; Mani, A.; Bera, A.K.; Maji, S.K.; Senapati, S.; Mullasari, A.S.; Mahapatra, N.R. Naturally occurring variants of the dysglycemic peptide pancreastatin: Differential potencies for multiple cellular functions and structure-function correlation. J. Biol. Chem. 2014, 289, 4455–4469.
- Krawczyk, S.A.; Haller, J.F.; Ferrante, T.; Zoeller, R.A.; Corkey, B.E. Reactive oxygen species facilitate translocation of hormone sensitive lipase to the lipid droplet during lipolysis in human differentiated adipocytes. PLoS ONE 2012, 7, e34904.
- Caturano, A.; D’Angelo, M.; Mormone, A.; Russo, V.; Mollica, M.P.; Salvatore, T.; Galiero, R.; Rinaldi, L.; Vetrano, E.; Marfella, R.; et al. Oxidative stress in type 2 diabetes: Impacts from pathogenesis to lifestyle modifications. Curr. Issues Mol. Biol. 2023, 45, 6651–6666.
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63.
- Rajan, S.; Shankar, K.; Beg, M.; Varshney, S.; Gupta, A.; Srivastava, A.; Kumar, D.; Mishra, R.K.; Hussain, Z.; Gayen, J.R.; et al. Chronic hyperinsulinemia reduces insulin sensitivity and metabolic functions of brown adipocyte. J. Endocrinol. 2016, 230, 275–290.
- Garg, R.; Gupta, A.P.; Katekar, R.; Verma, S.; Goand, U.K.; Dadge, S.; Gayen, J.R. Pancreastatin inhibitor psti8 prevents free fatty acid-induced oxidative stress and insulin resistance by modulating jnk pathway: In vitro and in vivo findings. Life Sci. 2022, 289, 120221.
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004, 24, 1844–1854.
- Mouche, S.; Mkaddem, S.B.; Wang, W.; Katic, M.; Tseng, Y.H.; Carnesecchi, S.; Steger, K.; Foti, M.; Meier, C.A.; Muzzin, P.; et al. Reduced expression of the NADPH oxidase nox4 is a hallmark of adipocyte differentiation. Biochim. Biophys. Acta 2007, 1773, 1015–1027.
- Schroder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245.
- Malik, S.A.; Acharya, J.D.; Mehendale, N.K.; Kamat, S.S.; Ghaskadbi, S.S. Pterostilbene reverses palmitic acid mediated insulin resistance in hepg2 cells by reducing oxidative stress and triglyceride accumulation. Free Radic. Res. 2019, 53, 815–827.
- Remsberg, C.M.; Yanez, J.A.; Ohgami, Y.; Vega-Villa, K.R.; Rimando, A.M.; Davies, N.M. Pharmacometrics of pterostilbene: Preclinical pharmacokinetics and metabolism, anticancer, antiinflammatory, antioxidant and analgesic activity. Phytother. Res. PTR 2008, 22, 169–179.
- Acharya, J.D.; Ghaskadbi, S.S. Protective effect of pterostilbene against free radical mediated oxidative damage. BMC Complement. Altern. Med. 2013, 13, 238.
- Satheesh, M.A.; Pari, L. Effect of pterostilbene on lipids and lipid profiles in streptozotocin-nicotinamide induced type 2 diabetes mellitus. J. Appl. Biomed. 2008, 6, 31–37.
- Elango, B.; Dornadula, S.; Paulmurugan, R.; Ramkumar, K.M. Pterostilbene ameliorates streptozotocin-induced diabetes through enhancing antioxidant signaling pathways mediated by nrf2. Chem. Res. Toxicol. 2016, 29, 47–57.
- Gomez-Zorita, S.; Fernandez-Quintela, A.; Aguirre, L.; Macarulla, M.T.; Rimando, A.M.; Portillo, M.P. Pterostilbene improves glycaemic control in rats fed an obesogenic diet: Involvement of skeletal muscle and liver. Food Funct. 2015, 6, 1968–1976.
- Pihlajaniemi, T.; Myllyla, R.; Kivirikko, K.I.; Tryggvason, K. Effects of streptozotocin diabetes, glucose, and insulin on the metabolism of type iv collagen and proteoglycan in murine basement membrane-forming ehs tumor tissue. J. Biol. Chem. 1982, 257, 14914–14920.
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620.
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N.T. Reactive oxygen species (ros) and wound healing: The functional role of ros and emerging ros-modulating technologies for augmentation of the healing process. Int. Wound J. 2017, 14, 89–96.
- Kim, Y.S.; Lee, H.Y.; Jang, J.Y.; Lee, H.R.; Shin, Y.S.; Kim, C.H. Redox treatment ameliorates diabetes mellitus-induced skin flap necrosis via inhibiting apoptosis and promoting neoangiogenesis. Exp. Biol. Med. 2021, 246, 718–728.
- Toyoda, K.; Fujii, K.; Kamouchi, M.; Nakane, H.; Arihiro, S.; Okada, Y.; Ibayashi, S.; Iida, M. Free radical scavenger, edaravone, in stroke with internal carotid artery occlusion. J. Neurol. Sci. 2004, 221, 11–17.
- Liu, J.; Zhang, D.; Luo, W.; Yu, J.; Li, J.; Yu, Y.; Zhang, X.; Chen, J.; Wu, X.R.; Huang, C. E3 ligase activity of xiap ring domain is required for xiap-mediated cancer cell migration, but not for its rhogdi binding activity. PLoS ONE 2012, 7, e35682.
- Nguyen, T.; Lau, D.C. The obesity epidemic and its impact on hypertension. Can. J. Cardiol. 2012, 28, 326–333.
- Franklin, S.S.; Gustin, W.t.; Wong, N.D.; Larson, M.G.; Weber, M.A.; Kannel, W.B.; Levy, D. Hemodynamic patterns of age-related changes in blood pressure. The framingham heart study. Circulation 1997, 96, 308–315.
- Pravenec, M.; Wallace, C.; Aitman, T.J.; Kurtz, T.W. Gene expression profiling in hypertension research: A critical perspective. Hypertension 2003, 41, 3–8.
- Radkowski, P.; Wator, G.; Skupien, J.; Bogdali, A.; Wolkow, P. Analysis of gene expression to predict dynamics of future hypertension incidence in type 2 diabetic patients. BMC Proc. 2016, 10, 113–117.
- Chen, G.; Adeyemo, A.A.; Zhou, J.; Chen, Y.; Doumatey, A.; Lashley, K.; Huang, H.; Amoah, A.; Agyenim-Boateng, K.; Eghan, B.A., Jr.; et al. A genome-wide search for linkage to renal function phenotypes in west africans with type 2 diabetes. Am. J. Kidney Dis. 2007, 49, 394–400.
- Kwak, S.H.; Hernandez-Cancela, R.B.; DiCorpo, D.A.; Condon, D.E.; Merino, J.; Wu, P.; Brody, J.A.; Yao, J.; Guo, X.; Ahmadizar, F.; et al. Time-to-event genome-wide association study for incident cardiovascular disease in people with type 2 diabetes mellitus. medRxiv 2023.
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645.
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415.
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761.
- Talior, I.; Yarkoni, M.; Bashan, N.; Eldar-Finkelman, H. Increased glucose uptake promotes oxidative stress and pkc-delta activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E295–E302.
- Wajngarten, M.; Silva, G.S. Hypertension and stroke: Update on treatment. Eur. Cardiol. 2019, 14, 111–115.
- Johansson, B.B. Hypertension mechanisms causing stroke. Clin. Exp. Pharmacol. Physiol. 1999, 26, 563–565.
- Neaton, J.D.; Blackburn, H.; Jacobs, D.; Kuller, L.; Lee, D.J.; Sherwin, R.; Shih, J.; Stamler, J.; Wentworth, D. Serum cholesterol level and mortality findings for men screened in the multiple risk factor intervention trial. Multiple risk factor intervention trial research group. Arch. Intern. Med. 1992, 152, 1490–1500.
- Peeling, J.; Yan, H.J.; Chen, S.G.; Campbell, M.; Del Bigio, M.R. Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res. 1998, 795, 63–70.
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786.
- Michihara, A.; Oda, A.; Mido, M. High expression levels of NADPH oxidase 3 in the cerebrum of ten-week-old stroke-prone spontaneously hypertensive rats. Biol. Pharm. Bull. 2016, 39, 252–258.
- Okamoto, K.; Yamamoto, K.; Morita, N.; Ohta, Y.; Chikugo, T.; Higashizawa, T.; Suzuki, T. Establishment and use of the m strain of stroke-prone spontaneously hypertensive rat. J. Hypertension. Suppl. 1986, 4, S21–S24.
- Iritani, N.; Fukuda, E.; Nara, Y.; Yamori, Y. Lipid metabolism in spontaneously hypertensive rats (shr). Atherosclerosis 1977, 28, 217–222.
- Tanito, M.; Nakamura, H.; Kwon, Y.W.; Teratani, A.; Masutani, H.; Shioji, K.; Kishimoto, C.; Ohira, A.; Horie, R.; Yodoi, J. Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxid. Redox Signal. 2004, 6, 89–97.
- Michihara, A.; Shimatani, M.; Anraku, M.; Tomida, H.; Akasaki, K. High levels of oxidative stress exist in the brain than serum or kidneys in stroke-prone spontaneously hypertensive rats at ten weeks of age. Biol. Pharm. Bull. 2010, 33, 518–521.
- Loft, S.; Vistisen, K.; Ewertz, M.; Tjonneland, A.; Overvad, K.; Poulsen, H.E. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: Influence of smoking, gender and body mass index. Carcinogenesis 1992, 13, 2241–2247.
- Nakamura, T.; Yamamoto, E.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Dong, Y.F.; Matsuba, S.; Ogawa, H.; Kim-Mitsuyama, S. Pioglitazone exerts protective effects against stroke in stroke-prone spontaneously hypertensive rats, independently of blood pressure. Stroke 2007, 38, 3016–3022.
- Ahmarani, L.; Avedanian, L.; Al-Khoury, J.; Perreault, C.; Jacques, D.; Bkaily, G. Whole-cell and nuclear NADPH oxidases levels and distribution in human endocardial endothelial, vascular smooth muscle, and vascular endothelial cells. Can. J. Physiol. Pharmacol. 2013, 91, 71–79.
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390.
- Nediani, C.; Borchi, E.; Giordano, C.; Baruzzo, S.; Ponziani, V.; Sebastiani, M.; Nassi, P.; Mugelli, A.; d’Amati, G.; Cerbai, E. NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and right ventricle. J. Mol. Cell Cardiol. 2007, 42, 826–834.
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171.
- Borchi, E.; Bargelli, V.; Stillitano, F.; Giordano, C.; Sebastiani, M.; Nassi, P.A.; d’Amati, G.; Cerbai, E.; Nediani, C. Enhanced ros production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim. Biophys. Acta 2010, 1802, 331–338.
- Maack, C.; Kartes, T.; Kilter, H.; Schafers, H.J.; Nickenig, G.; Bohm, M.; Laufs, U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-gtpase and represents a target for statin treatment. Circulation 2003, 108, 1567–1574.
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041.
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH oxidase nox4 drives cardiac differentiation: Role in regulating cardiac transcription factors and map kinase activation. Mol. Biol. Cell 2006, 17, 3978–3988.
- Fenyo, I.M.; Florea, I.C.; Raicu, M.; Manea, A. Tyrphostin ag490 reduces napdh oxidase activity and expression in the aorta of hypercholesterolemic apolipoprotein e-deficient mice. Vasc. Pharmacol. 2011, 54, 100–106.
- Sirker, A.; Murdoch, C.E.; Protti, A.; Sawyer, G.J.; Santos, C.X.; Martin, D.; Zhang, X.; Brewer, A.C.; Zhang, M.; Shah, A.M. Cell-specific effects of nox2 on the acute and chronic response to myocardial infarction. J. Mol. Cell Cardiol. 2016, 98, 11–17.
- Bell, R.M.; Cave, A.C.; Johar, S.; Hearse, D.J.; Shah, A.M.; Shattock, M.J. Pivotal role of nox-2-containing NADPH oxidase in early ischemic preconditioning. FASEB J. 2005, 19, 2037–2039.
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: Reality or just an attractive hypothesis? Trends Neurosci. 2008, 31, 251–256.
- Wind, S.; Beuerlein, K.; Armitage, M.E.; Taye, A.; Kumar, A.H.; Janowitz, D.; Neff, C.; Shah, A.M.; Wingler, K.; Schmidt, H.H. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by nox1/2 is reversed by NADPH oxidase inhibition. Hypertension 2010, 56, 490–497.
- Bkaily, G.; Najibeddine, W.; Jacques, D. Increase of NADPH oxidase 3 in heart failure of hereditary cardiomyopathy (1). Can. J. Physiol. Pharmacol. 2019, 97, 902–908.
- Dhalla, A.K.; Hill, M.F.; Singal, P.K. Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 1996, 28, 506–514.
- Wu, R.; Yin, D.; Sadekova, N.; Deschepper, C.F.; de Champlain, J.; Girouard, H. Protective effects of aspirin from cardiac hypertrophy and oxidative stress in cardiomyopathic hamsters. Oxidative Med. Cell. Longev. 2012, 2012, 761710.
- Mollnau, H.; Oelze, M.; August, M.; Wendt, M.; Daiber, A.; Schulz, E.; Baldus, S.; Kleschyov, A.L.; Materne, A.; Wenzel, P.; et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2554–2559.
- Jacques, D.; Bkaily, G. Endocardial endothelial cell hypertrophy takes place during the development of hereditary cardiomyopathy. Mol. Cell Biochem. 2019, 453, 157–161.
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456.
- Bolli, R.; Jeroudi, M.O.; Patel, B.S.; Aruoma, O.I.; Halliwell, B.; Lai, E.K.; McCay, P.B. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ. Res. 1989, 65, 607–622.
- Zhao, Z.Q.; Nakamura, M.; Wang, N.P.; Wilcox, J.N.; Shearer, S.; Ronson, R.S.; Guyton, R.A.; Vinten-Johansen, J. Reperfusion induces myocardial apoptotic cell death. Cardiovasc. Res. 2000, 45, 651–660.
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47.
- Kloner, R.A.; Jennings, R.B. Consequences of brief ischemia: Stunning, preconditioning, and their clinical implications: Part 2. Circulation 2001, 104, 3158–3167.
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Ikeuchi, M.; Matsusaka, H.; Suematsu, N.; Wen, J.; Egashira, K.; Takeshita, A. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2003, 108, 2134–2140.
- Kaikita, K.; Hayasaki, T.; Okuma, T.; Kuziel, W.A.; Ogawa, H.; Takeya, M. Targeted deletion of cc chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004, 165, 439–447.
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. Ccl2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889.
- Morimoto, H.; Hirose, M.; Takahashi, M.; Kawaguchi, M.; Ise, H.; Kolattukudy, P.E.; Yamada, M.; Ikeda, U. Mcp-1 induces cardioprotection against ischaemia/reperfusion injury: Role of reactive oxygen species. Cardiovasc. Res. 2008, 78, 554–562.
- Kolattukudy, P.E.; Quach, T.; Bergese, S.; Breckenridge, S.; Hensley, J.; Altschuld, R.; Gordillo, G.; Klenotic, S.; Orosz, C.; Parker-Thornburg, J. Myocarditis induced by targeted expression of the mcp-1 gene in murine cardiac muscle. Am. J. Pathol. 1998, 152, 101–111.
- Vats, S.; Sundquist, K.; Sundquist, J.; Zhang, N.; Wang, X.; Acosta, S.; Gottsater, A.; Memon, A.A. Oxidative stress-related genetic variation and antioxidant vitamin intake in intact and ruptured abdominal aortic aneurysm: A swedish population-based retrospective cohort study. Eur. J. Prev. Cardiol. 2024, 31, 61–74.
- Manjer, J.; Elmstahl, S.; Janzon, L.; Berglund, G. Invitation to a population-based cohort study: Differences between subjects recruited using various strategies. Scand. J. Public Health 2002, 30, 103–112.
- Wanhainen, A.; Verzini, F.; Van Herzeele, I.; Allaire, E.; Bown, M.; Cohnert, T.; Dick, F.; van Herwaarden, J.; Karkos, C.; Koelemay, M.; et al. Editor’s choice—European society for vascular surgery (ESVS) 2019 clinical practice guidelines on the management of abdominal aorto-iliac artery aneurysms. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 8–93.
- Howard, D.P.; Banerjee, A.; Fairhead, J.F.; Handa, A.; Silver, L.E.; Rothwell, P.M.; Oxford Vascular, S. Age-specific incidence, risk factors and outcome of acute abdominal aortic aneurysms in a defined population. Br. J. Surg. 2015, 102, 907–915.
- Reimerink, J.J.; van der Laan, M.J.; Koelemay, M.J.; Balm, R.; Legemate, D.A. Systematic review and meta-analysis of population-based mortality from ruptured abdominal aortic aneurysm. Br. J. Surg. 2013, 100, 1405–1413.
- Song, P.; He, Y.; Adeloye, D.; Zhu, Y.; Ye, X.; Yi, Q.; Rahimi, K.; Rudan, I.; Global Health Epidemiology Research Group. The global and regional prevalence of abdominal aortic aneurysms: A systematic review and modeling analysis. Ann. Surg. 2023, 277, 912–919.
- Sweeting, M.J.; Thompson, S.G.; Brown, L.C.; Powell, J.T.; Collaborators, R. Meta-analysis of individual patient data to examine factors affecting growth and rupture of small abdominal aortic aneurysms. Br. J. Surg. 2012, 99, 655–665.
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwala, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Zmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms--association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396.
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 461–469.
- Kim, H.W.; Blomkalns, A.L.; Ogbi, M.; Thomas, M.; Gavrila, D.; Neltner, B.S.; Cassis, L.A.; Thompson, R.W.; Weiss, R.M.; Lindower, P.D.; et al. Role of myeloperoxidase in abdominal aortic aneurysm formation: Mitigation by taurine. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1168–H1179.
- Ludvigsson, J.F.; Andersson, E.; Ekbom, A.; Feychting, M.; Kim, J.L.; Reuterwall, C.; Heurgren, M.; Olausson, P.O. External review and validation of the swedish national inpatient register. BMC Public Health 2011, 11, 450.
- Caramori, M.L.; Fioretto, P.; Mauer, M. Low glomerular filtration rate in normoalbuminuric type 1 diabetic patients: An indicator of more advanced glomerular lesions. Diabetes 2003, 52, 1036–1040.
- Cockcroft, D.W.; Gault, M.H. Prediction of creatinine clearance from serum creatinine. Nephron 1976, 16, 31–41.
- Hunt, S.C.; Hasstedt, S.J.; Coon, H.; Camp, N.J.; Cawthon, R.M.; Wu, L.L.; Hopkins, P.N. Linkage of creatinine clearance to chromosome 10 in utah pedigrees replicates a locus for end-stage renal disease in humans and renal failure in the fawn-hooded rat. Kidney Int. 2002, 62, 1143–1148.
- Ye, S.; Zhong, H.; Yanamadala, S.; Campese, V.M. Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension 2006, 48, 309–315.
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82.
- Rokutan, K.; Kawahara, T.; Kuwano, Y.; Tominaga, K.; Nishida, K.; Teshima-Kondo, S. Nox enzymes and oxidative stress in the immunopathology of the gastrointestinal tract. Semin. Immunopathol. 2008, 30, 315–327.
- Banfi, B.; Maturana, A.; Jaconi, S.; Arnaudeau, S.; Laforge, T.; Sinha, B.; Ligeti, E.; Demaurex, N.; Krause, K.H. A mammalian h+ channel generated through alternative splicing of the NADPH oxidase homolog noh-1. Science 2000, 287, 138–142.
- Teshima, S.; Kutsumi, H.; Kawahara, T.; Kishi, K.; Rokutan, K. Regulation of growth and apoptosis of cultured guinea pig gastric mucosal cells by mitogenic oxidase 1. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1169–G1176.
- Salles, N.; Szanto, I.; Herrmann, F.; Armenian, B.; Stumm, M.; Stauffer, E.; Michel, J.P.; Krause, K.H. Expression of mrna for ros-generating NADPH oxidases in the aging stomach. Exp. Gerontol. 2005, 40, 353–357.
- Augusto, A.C.; Miguel, F.; Mendonca, S.; Pedrazzoli, J., Jr.; Gurgueira, S.A. Oxidative stress expression status associated to Helicobacter pylori virulence in gastric diseases. Clin. Biochem. 2007, 40, 615–622.
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504.
- Dupuy, C.; Pomerance, M.; Ohayon, R.; Noel-Hudson, M.S.; Deme, D.; Chaaraoui, M.; Francon, J.; Virion, A. Thyroid oxidase (thox2) gene expression in the rat thyroid cell line frtl-5. Biochem. Biophys. Res. Commun. 2000, 277, 287–292.
- El Hassani, R.A.; Benfares, N.; Caillou, B.; Talbot, M.; Sabourin, J.C.; Belotte, V.; Morand, S.; Gnidehou, S.; Agnandji, D.; Ohayon, R.; et al. Dual oxidase2 is expressed all along the digestive tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G933–G942.
- Plantinga, T.S.; Arts, P.; Knarren, G.H.; Mulder, A.H.; Wakelkamp, I.M.; Hermus, A.R.; Joosten, L.A.; Netea, M.G.; Bisschop, P.H.; de Herder, W.W.; et al. Rare nox3 variants confer susceptibility to agranulocytosis during thyrostatic treatment of graves’ disease. Clin. Pharmacol. Ther. 2017, 102, 1017–1024.
- Yang, J.; Zhong, J.; Xiao, X.H.; Zhou, L.Z.; Chen, Y.J.; Liu, J.H.; Cao, R.X.; Wen, G.B. The relationship between bone marrow characteristics and the clinical prognosis of antithyroid drug-induced agranulocytosis. Endocr. J. 2013, 60, 185–189.
- Ishimaru, N.; Ohnishi, H.; Nishiuma, T.; Doukuni, R.; Umezawa, K.; Oozone, S.; Kuramoto, E.; Yoshimura, S.; Kinami, S. Antithyroid drug-induced agranulocytosis complicated by pneumococcal sepsis and upper airway obstruction. Intern. Med. 2013, 52, 2355–2359.
- Kubota, Y.; Toh Yoon, E.W. Severe drug-induced agranulocytosis successfully treated with recombinant human granulocyte colony-stimulating factor. Case Rep. Med. 2018, 2018, 8439791.
- Andrès, E.; Maloisel, F. Antibiotic-induced agranulocytosis: A monocentric study of 21 cases. Arch. Intern. Med. 2001, 161, 2619.
- di Fonzo, H.; Villegas Gutsch, M.; Castroagudin, A.; Cabrera, M.V.; Mazzei, M.E.; Rueda, D. Agranulocytosis induced by vancomycin. Case report and literature review. Am. J. Case Rep. 2018, 19, 1053–1056.
- Weissel, M. Propylthiouracil: Clinical overview of its efficacy and its side effects more than 50 years after the introduction of its use in thyrostatic treatment. Exp. Clin. Endocrinol. Diabetes 2010, 118, 101–104.
- Sato, S.; Noh, J.Y.; Sato, S.; Suzuki, M.; Yasuda, S.; Matsumoto, M.; Kunii, Y.; Mukasa, K.; Sugino, K.; Ito, K.; et al. Comparison of efficacy and adverse effects between methimazole 15 mg+inorganic iodine 38 mg/day and methimazole 30 mg/day as initial therapy for graves’ disease patients with moderate to severe hyperthyroidism. Thyroid 2015, 25, 43–50.
- Taylor, P.N.; Vaidya, B. Side effects of anti-thyroid drugs and their impact on the choice of treatment for thyrotoxicosis in pregnancy. Eur. Thyroid J. 2012, 1, 176–185.
- Andersohn, F.; Konzen, C.; Garbe, E. Systematic review: Agranulocytosis induced by nonchemotherapy drugs. Ann. Intern. Med. 2007, 146, 657–665.
- Takata, K.; Kubota, S.; Fukata, S.; Kudo, T.; Nishihara, E.; Ito, M.; Amino, N.; Miyauchi, A. Methimazole-induced agranulocytosis in patients with graves’ disease is more frequent with an initial dose of 30 mg daily than with 15 mg daily. Thyroid 2009, 19, 559–563.
- Chu, X.; Pan, C.M.; Zhao, S.X.; Liang, J.; Gao, G.Q.; Zhang, X.M.; Yuan, G.Y.; Li, C.G.; Xue, L.Q.; Shen, M.; et al. A genome-wide association study identifies two new risk loci for graves’ disease. Nat. Genet. 2011, 43, 897–901.
- Liu, W.; Wang, H.N.; Gu, Z.H.; Yang, S.Y.; Ye, X.P.; Pan, C.M.; Zhao, S.X.; Xue, L.Q.; Xie, H.J.; Yu, S.S.; et al. Identification of bach2 as a susceptibility gene for graves’ disease in the chinese han population based on a three-stage genome-wide association study. Hum. Genet. 2014, 133, 661–671.
- Roderique, J.D.; Josef, C.S.; Feldman, M.J.; Spiess, B.D. A modern literature review of carbon monoxide poisoning theories, therapies, and potential targets for therapy advancement. Toxicology 2015, 334, 45–58.
- Varma, D.R.; Mulay, S.; Chemtob, S. Chapter 20—Carbon monoxide: From public health risk to painless killer. In Handbook of Toxicology of Chemical Warfare Agents; Gupta, R.C., Ed.; Academic Press: San Diego, CA, USA, 2009; pp. 271–292.
- Ginsberg, M.D. Delayed neurological deterioration following hypoxia. Adv. Neurol. 1979, 26, 21–44.
- Hara, S.; Mizukami, H.; Kurosaki, K.; Kuriiwa, F.; Mukai, T. Existence of a threshold for hydroxyl radical generation independent of hypoxia in rat striatum during carbon monoxide poisoning. Arch. Toxicol. 2011, 85, 1091–1099.
- Sun, Q.; Cai, J.; Zhou, J.; Tao, H.; Zhang, J.H.; Zhang, W.; Sun, X.J. Hydrogen-rich saline reduces delayed neurologic sequelae in experimental carbon monoxide toxicity. Crit. Care Med. 2011, 39, 765–769.
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694.
- Piantadosi, C.A.; Zhang, J.; Demchenko, I.T. Production of hydroxyl radical in the hippocampus after co hypoxia or hypoxic hypoxia in the rat. Free Radic. Biol. Med. 1997, 22, 725–732.
- Hara, S.; Kobayashi, M.; Kuriiwa, F.; Mukai, T.; Mizukami, H. Different mechanisms of hydroxyl radical production susceptible to purine p2 receptor antagonists between carbon monoxide poisoning and exogenous atp in rat striatum. Free Radic. Res. 2014, 48, 1322–1333.
- Hara, S.; Kobayash, M.; Kuriiwa, F.; Kurosaki, K.; Mizukami, H. Gene expression in rat striatum following carbon monoxide poisoning. Genom Data 2017, 12, 74–75.
- Hara, S.; Kobayashi, M.; Kuriiwa, F.; Ikematsu, K.; Mizukami, H. Hydroxyl radical production via NADPH oxidase in rat striatum due to carbon monoxide poisoning. Toxicology 2018, 394, 63–71.
- Thom, S.R.; Ohnishi, S.T.; Ischiropoulos, H. Nitric oxide released by platelets inhibits neutrophil b2 integrin function following acute carbon monoxide poisoning. Toxicol. Appl. Pharmacol. 1994, 128, 105–110.
- Nabeshima, T.; Katoh, A.; Ishimaru, H.; Yoneda, Y.; Ogita, K.; Murase, K.; Ohtsuka, H.; Inari, K.; Fukuta, T.; Kameyama, T. Carbon monoxide-induced delayed amnesia, delayed neuronal death and change in acetylcholine concentration in mice. J. Pharmacol. Exp. Ther. 1991, 256, 378–384.
- Fujita, K.; Seike, T.; Yutsudo, N.; Ohno, M.; Yamada, H.; Yamaguchi, H.; Sakumi, K.; Yamakawa, Y.; Kido, M.A.; Takaki, A.; et al. Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of parkinson’s disease. PLoS ONE 2009, 4, e7247.
- Mikkola, L.; Holopainen, S.; Pessa-Morikawa, T.; Lappalainen, A.K.; Hytonen, M.K.; Lohi, H.; Iivanainen, A. Genetic dissection of canine hip dysplasia phenotypes and osteoarthritis reveals three novel loci. BMC Genom. 2019, 20, 1027.
- Skurkova, L.; Hluchy, M.; Lackova, M.; Mihalova, M.; Ledecky, V. Relation of the norberg angle and position of the femoral head centre to the dorsal acetabular edge in evaluation of canine hip dysplasia. Vet. Comp. Orthop. Traumatol. 2010, 23, 433–438.
- Broeckx, B.J.G.; Vezzoni, A.; Bogaerts, E.; Bertal, M.; Bosmans, T.; Stock, E.; Deforce, D.; Peelman, L.; Saunders, J.H. Comparison of three methods to quantify laxity in the canine hip joint. Vet. Comp. Orthop. Traumatol. V.C.O.T 2018, 31, 23–29.
- Sanchez-Molano, E.; Woolliams, J.A.; Pong-Wong, R.; Clements, D.N.; Blott, S.C.; Wiener, P. Quantitative trait loci mapping for canine hip dysplasia and its related traits in uk labrador retrievers. BMC Genom. 2014, 15, 833.
- Lavrijsen, I.C.; Leegwater, P.A.; Martin, A.J.; Harris, S.J.; Tryfonidou, M.A.; Heuven, H.C.; Hazewinkel, H.A. Genome wide analysis indicates genes for basement membrane and cartilage matrix proteins as candidates for hip dysplasia in labrador retrievers. PLoS ONE 2014, 9, e87735.
- Fels, L.; Distl, O. Identification and validation of quantitative trait loci (qtl) for canine hip dysplasia (chd) in german shepherd dogs. PLoS ONE 2014, 9, e96618.
- Pfahler, S.; Distl, O. Identification of quantitative trait loci (qtl) for canine hip dysplasia and canine elbow dysplasia in bernese mountain dogs. PLoS ONE 2012, 7, e49782.
- Zhou, Z.; Sheng, X.; Zhang, Z.; Zhao, K.; Zhu, L.; Guo, G.; Friedenberg, S.G.; Hunter, L.S.; Vandenberg-Foels, W.S.; Hornbuckle, W.E.; et al. Differential genetic regulation of canine hip dysplasia and osteoarthritis. PLoS ONE 2010, 5, e13219.
- Carlstrom, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ros pathway in monocytes. Nat. Commun. 2019, 10, 3081.
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the phase 3 define and confirm studies. Mult. Scler. 2017, 23, 1875–1883.
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107.
- Kraaij, M.D.; Savage, N.D.; van der Kooij, S.W.; Koekkoek, K.; Wang, J.; van den Berg, J.M.; Ottenhoff, T.H.; Kuijpers, T.W.; Holmdahl, R.; van Kooten, C.; et al. Induction of regulatory t cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691.
- George-Chandy, A.; Nordstrom, I.; Nygren, E.; Jonsson, I.M.; Postigo, J.; Collins, L.V.; Eriksson, K. Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur. J. Immunol. 2008, 38, 1118–1126.
- Mossberg, N.; Movitz, C.; Hellstrand, K.; Bergstrom, T.; Nilsson, S.; Andersen, O. Oxygen radical production in leukocytes and disease severity in multiple sclerosis. J. Neuroimmunol. 2009, 213, 131–134.
- Mix, E.; Stefan, K.; Hoppner, J.; Klauer, T.; Zettl, U.K.; Strauss, U.; Meyer-Rienecker, H.J.; Rolfs, A. Lymphocyte subpopulations, oxidative burst and apoptosis in peripheral blood cells of patients with multiple sclerosis-effect of interferon-beta. Autoimmunity 2003, 36, 291–305.
- Becanovic, K.; Jagodic, M.; Sheng, J.R.; Dahlman, I.; Aboul-Enein, F.; Wallstrom, E.; Olofsson, P.; Holmdahl, R.; Lassmann, H.; Olsson, T. Advanced intercross line mapping of eae5 reveals ncf-1 and cldn4 as candidate genes for experimental autoimmune encephalomyelitis. J. Immunol. 2006, 176, 6055–6064.
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the nrf2 antioxidant pathway. Brain A J. Neurol. 2011, 134, 678–692.
- Li, L.; Sheng, Q.; Zeng, H.; Li, W.; Wang, Q.; Ma, G.; Xu, X.; Qiu, M.; Zhang, W.; Shan, C. Specific genetic aberrations of parathyroid in chinese patients with tertiary hyperparathyroidism using whole-exome sequencing. Front. Endocrinol. 2023, 14, 1221060.
- Messa, P.; Alfieri, C.M. Secondary and tertiary hyperparathyroidism. Front. Horm. Res. 2019, 51, 91–108.
- Alfieri, C.; Mattinzoli, D.; Messa, P. Tertiary and postrenal transplantation hyperparathyroidism. Endocrinol. Metab. Clin. North Am. 2021, 50, 649–662.
- Dream, S.; Chen, H.; Lindeman, B. Tertiary hyperparathyroidism: Why the delay? Ann. Surg. 2021, 273, e120–e122.
More