Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Francesco Nappi.
The use of next-generation sequencing has provided new insights into the causes and mechanisms of congenital heart disease (CHD). Examinations of the whole exome sequence have detected detrimental gene variations modifying single or contiguous nucleotides, which are characterised as pathogenic based on statistical assessments of families and correlations with congenital heart disease, elevated expression during heart development, and reductions in harmful protein-coding mutations in the general population.
- congenital heart disease
- first heart field
- second heart field
- whole-exome sequencing
- whole-genome sequencing
1. Current Knowledge
Congenital heart disease is widely acknowledged as the most frequent and frequently serious abnormality present at birth, with a prevalence of 6–13 per 1000 newborn infants [1,2,3,4,5,6][1][2][3][4][5][6]. CHD encompasses a broad spectrum of heart abnormalities, ranging from a single defect, such as an atrial septal defect (ASD) or a ventricular septal defect (VSD), or an isolated dysplastic valve, to more complex disorders. Cardiac conditions such as tetralogy of Fallot (TOF) or hypoplastic left heart syndrome (HLHS) are characterised by multiple defects. Figure 1 and Figure 2 show the improved operative and management techniques that have led to a significant reduction in the mortality rate of CHD. These techniques are used to treat critical malformations that require early intervention for survival. Over 90% of CHD patients survive into adulthood [7], resulting in a higher prevalence of CHD in the general population. In addition, the extended lifespan of patients with CHD has prompted greater acknowledgement of the related comorbidities. Childhood extracardiac structural or functional abnormalities and neurodevelopmental delays occur in approximately 13% of newborns with CHD [3,8][3][8]. CHD is considered syndromic if other diagnoses are likely to have resulted from the same cause. Syndromic CHD often results in complex heart malformations that can cause lifelong health problems affecting various bodily systems.
Therefore, an understanding of the causes and underlying pathways of CHD can provide valuable information about both the healthy and abnormal development of multiple organs.
Genetics
Due to significant technical advances in human genome research, genetics has now been found to play a crucial role in the development of CHD. Evidence initially emerged from examining patients with syndromic CHD through karyotyping, which identified aneuploidies such as trisomy 13, 18, or 21 (Down syndrome) and monosomy X (Turner syndrome) in approximately 12% of CHD patients [9]. Cytogenetic analyses and genomic arrays have aided in the understanding of subchromosomal structural rearrangements. These analyses have highlighted frequent 3 Mb deletions, which are also associated with DiGeorge (velocardiofacial) syndrome [10,11,12,13][10][11][12][13]. Deletions of 22q11.2, as determined by fluorescence in situ hybridisation (FISH) [14,15][14][15] and targeted amplification [16], are present in approximately 2% of all cases of congenital heart disease and in 13% of individuals with selected heart malformations. About 25% of sporadic CHD cases are caused by the coexistence of karyotype or microarray-detected abnormalities [17,18][17][18]. The preliminary detection of the monogenic causes of familial CHD has been facilitated by genome-wide linkage causes of familial CHD, encompassing variations in genes encoding transcription factors and transcriptional regulators of genes involved in heart growth [19,20][19][20]. Enhanced comprehension of the human genome at the base pair level, along with advancements in sequencing technologies, has facilitated the identification of genes linked to CHD. Harmful variants, such as missense mutations, loss-of-function mutations (LOFs), small insertions or deletions, copy number variants (CNVs), and structural defects, can be identified using advanced whole-exome sequencing (WES) and whole-genome sequencing (WGS) platforms, along with bioinformatic tools and large sequencing datasets from population-based studies. These techniques have been used to identify harmful variants present in individuals with a family history of congenital heart disease, new mutations in CHD subjects with healthy parents, and a significantly higher frequency of mutations in approximately 20% of CHD patients in large-cohort studies [18,19,20,21,22,23,24,25][18][19][20][21][22][23][24][25].
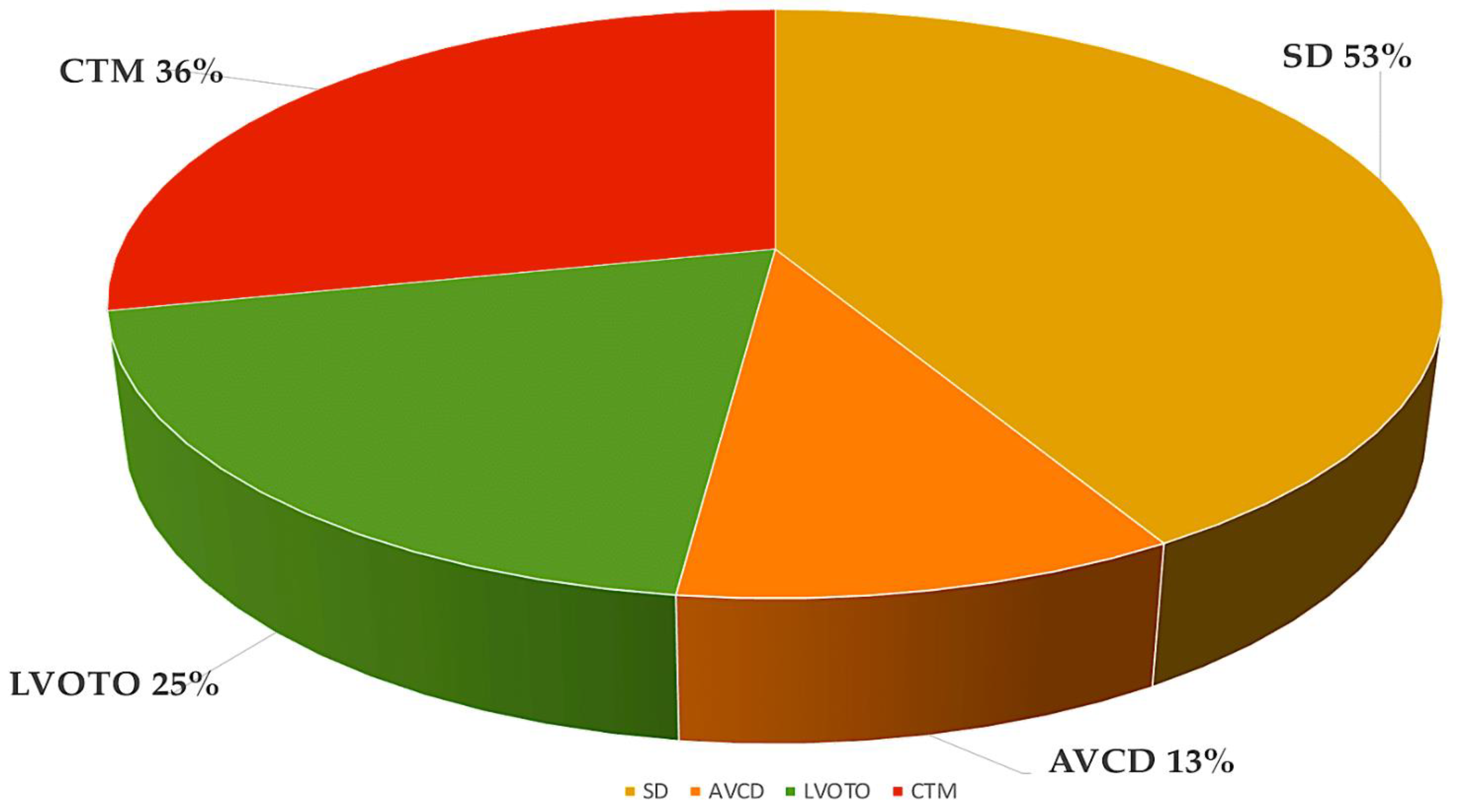
Figure 1. The most frequent forms of CHD from the Paediatric Cardiac Genomics Consortium, with percentages indicating the prevalence of the malformation in the patient population. It is important to note that the percentages exceed 100% as a result of concomitant structural cardiac abnormalities in the individuals Abbreviations: AVCD, atrioventricular canal defect; CHD, congenital heart disease; CTM, conotruncal malformation; LVOTO, left ventricular outflow tract obstruction; SD, septal defect.
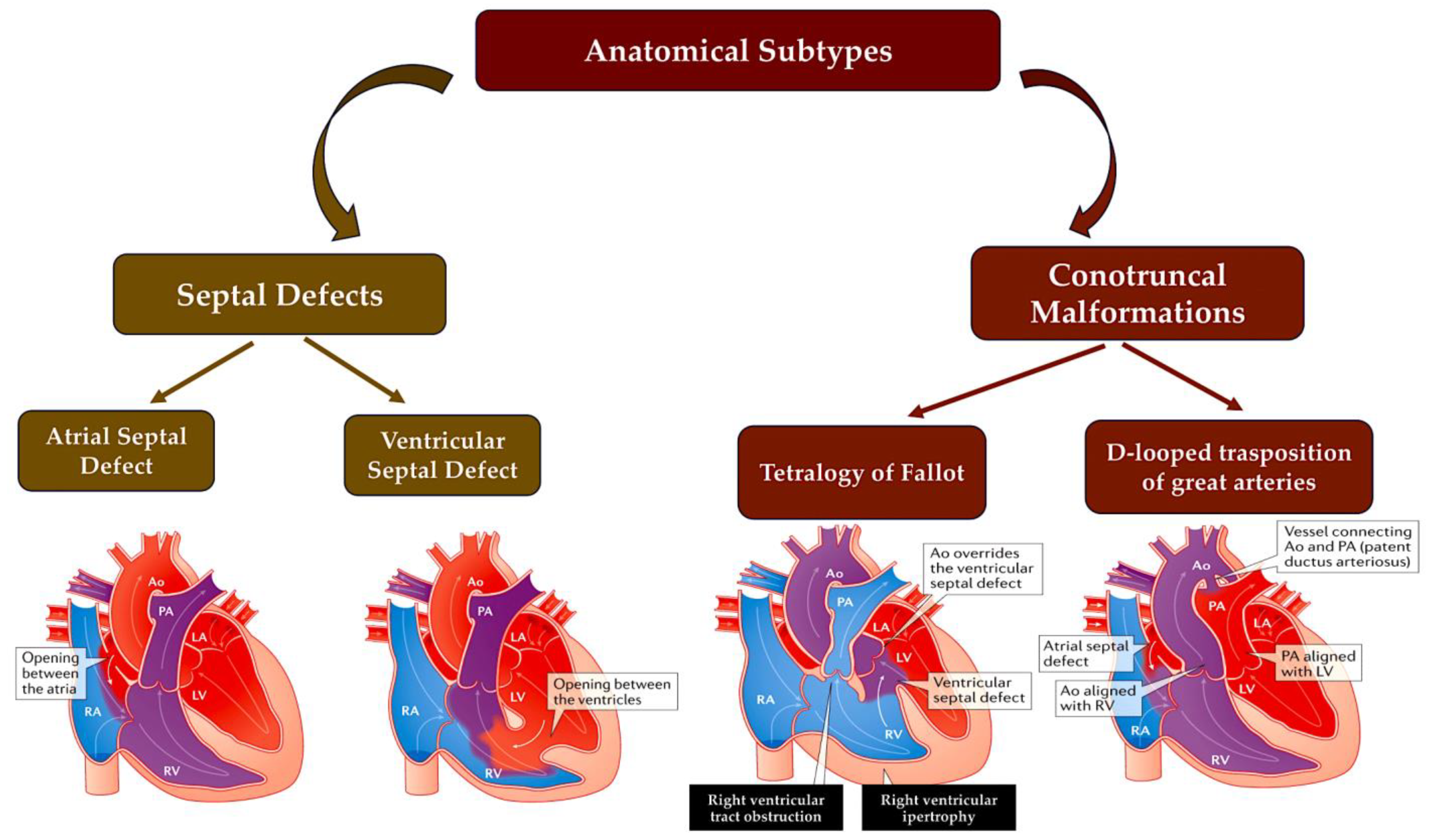
Figure 2. A simplified scheme of CHD defects divided according to anatomical subtype and their corresponding oxygen saturation levels. Anomalies in the communication between the left and right ventricles, such as ASD or VSD or atrioventricular canal defects, usually result in acyanotic states due to a left-to-right shunting of oxygen-rich blood into the pulmonary circulation. LVOTOs can range from isolated aortic stenosis to a combination of anomalies, such as hypoplastic left heart syndrome with mitral and aortic stenosis, which can cause cyanosis. Conotruncal malformations, such as TOF and TGV, are often associated with cyanosis due to the shunting of deoxygenated blood into the systemic circulation. This information is from the Paediatric Cardiac Genomics Consortium. Abbreviations: Ao, aorta; LA, left atrium; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RV, right ventricle; TGV, transposition of great vessel; TOF, tetralogy of Fallot.
In 45% of patients with CHD, these methods can detect harmful coding mutations in genes that are definitively or potentially linked to CHD [18,19,20,21,22,23,24,25][18][19][20][21][22][23][24][25]. Genes that are strongly associated with CHD exhibit mutations that significantly co-segregate within families affected by CHD, are significantly more prevalent in unrelated patients with CHD compared to control groups, or cause heart defects in individuals with CHD linked to syndromes. CHD candidate genes share common features, but their properties lack statistical significance for definitive categorisation. Different types of variants, such as aneuploidies, copy number variants, structural genetic variations, and LOF variations, affect the dosage of genes related to CHD. However, deleterious missense variants can preserve the physiologic gene dosage whilst impairing the function of the encoded protein [21,24][21][24]. Please refer to Figure 3.
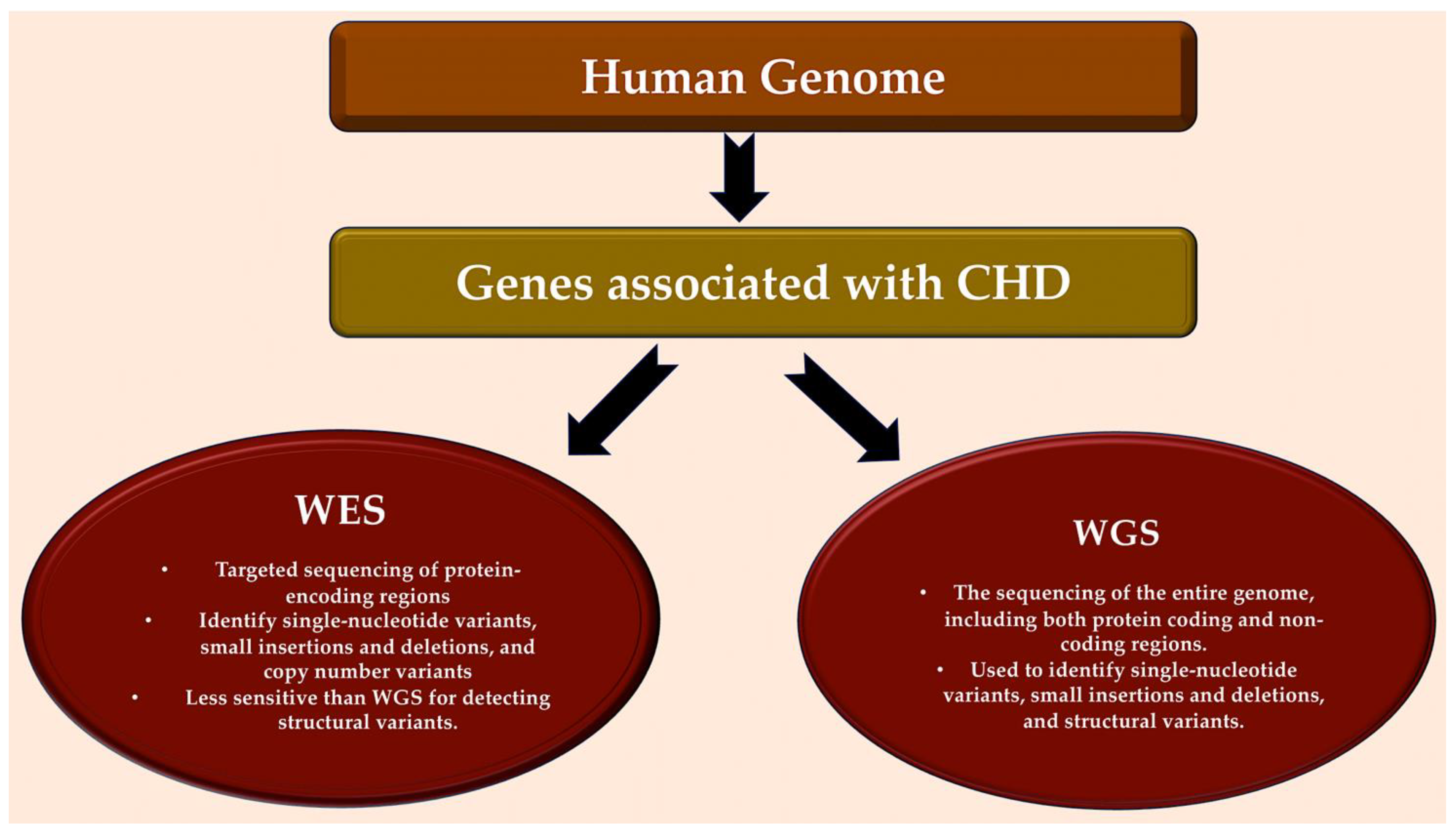
Figure 3. Detection of genes associated with CHD in human genome. Bioinformatic analytical tools such as WES and WGS platforms can be used to identify deleterious missense and LOF variants, small insertions or deletions, CNVs, and structural variants in large, aggregated, population-based sequencing datasets. Abbreviations: CHD, congenital heart disease; CNV, copy number variant; LOF, loss-of-function; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Genes linked to CHD provide new knowledge about the essential regulatory molecules and pathways involved in cardiogenesis, complementing and extending the wealth of information gained from studying heart development in experimental models. Studying spontaneous gene variants in sporadic CHD can reveal novel genes implicated in cardiogenesis while providing detailed information [17,21,24][17][21][24]. Clinical evaluations enable the identification of a wider range of cardiac and extracardiac phenotypes than studies conducted on experimental models. Most human variants have a heterozygous dosage, and while experimental models usually examine homozygous variants, data on congenital heart disease obtained from human studies can reveal information about less obvious impacts on heart formation. Furthermore, patients with critical and complex malformations display the greatest frequency of de novo damaging variants in genes linked to CHD, which provides convincing evidence of severe adverse effects on reproductive health and the evolutionary restriction on numerous genes connected with CHD [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25].
The question of why CHD is the most prevalent congenital abnormality is intriguing. One hypothesis suggests that the intricate developmental processes involved in heart formation are highly responsive to alterations in gene dosage for many essential genes and pathways. Such changes can cause developmental errors, many of which can be readily detected by the 18-week gestational age screening ultrasonography [26,27][26][27]. To showcase the intricate molecular orchestration of heart formation, thires reviewearchers provides a brief summary of cardiac development. Following this, the latest genomic findings and approaches that have been at the forefront of current CHD science over the last decade are discussed. Key signals that govern cardiac morphogenesis have been extensively examined in several informative reviews, which are recommended for further details, including publications [28,29,30,31,32,33,34,35][28][29][30][31][32][33][34][35]. Genes with harmful variants found in people with CHD are highlighted in bold during the report (Figure 4, [22,23,36,37,38,39,40][22][23][36][37][38][39][40]).
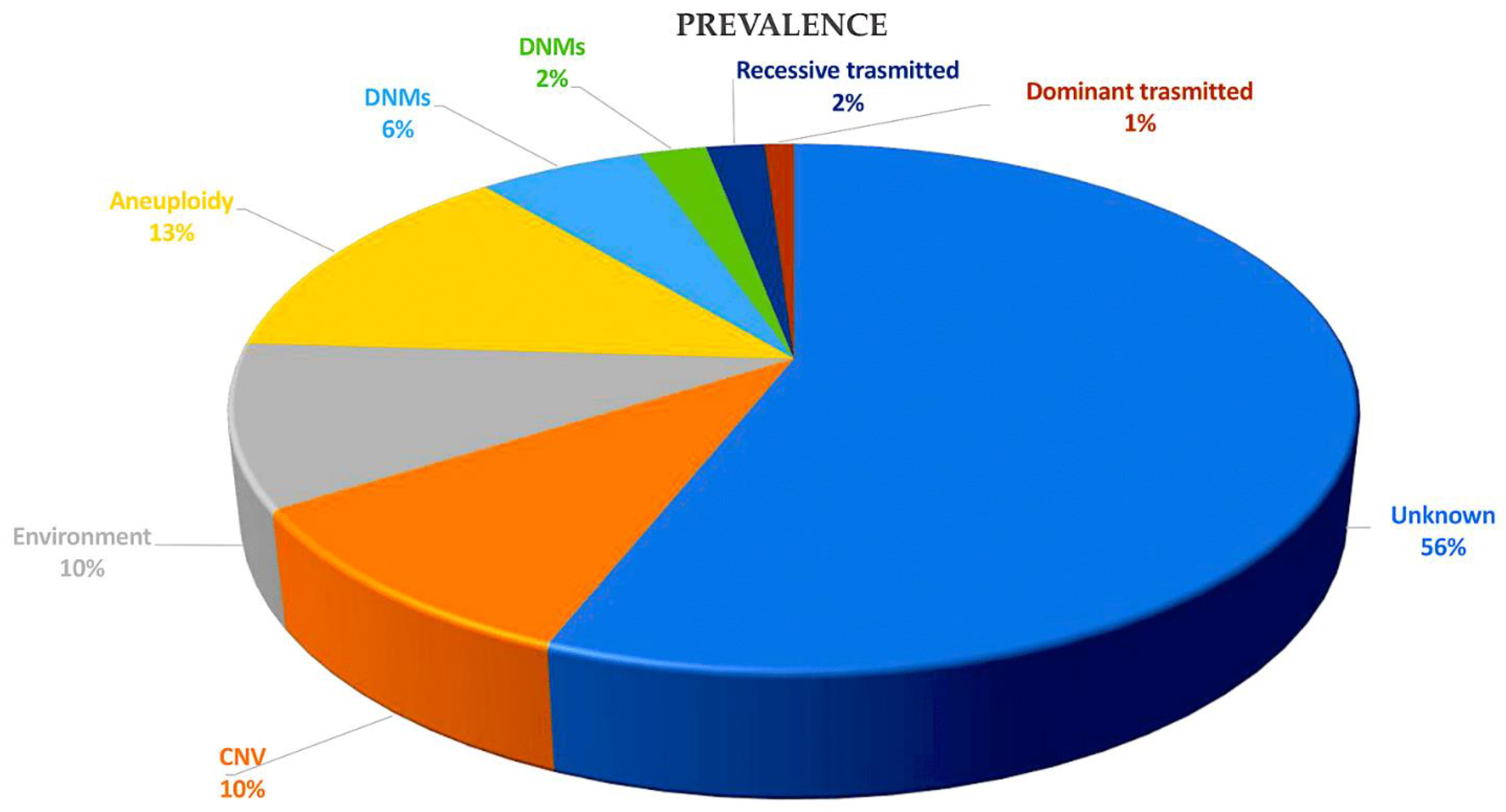
Figure 4. This pie chart illustrates the various causes of CHD and their relative proportions in contributing to the disease. Recessive transmitted causes involve variants in cilia and known CHD genes. Dominant causes of TOF include LOFs associated with FLT4, KDR, TBX1, and NOTCH1. DNMs may involve chromatin modifiers (2%) and dominant CHD genes (6%). Aneuploidy aetiology is related to trisomy 13, 18, or 21 and monosomy X. Non-genetic causes of CHD include teratogenic exposures such as alcohol; antiseizure and antiretroviral medication; and illnesses and infectious agents, such as diabetes, hypercholesterolemia, and rubella. The causes of CHD are investigated through gene–gene interactions, gene–environment interactions, polygenic inheritance, and epigenetics. CNVs, including Del22q11.2, Del20p12, Del17q11, Del11q24-25, Del8p23.1, Del1q21.1, Dup12q24, and Dup7q11.23, are also considered. Abbreviations: CHD, congenital heart disease; CNVs, copy number variations; Del, deletion, DMSs, de novo mutations; Dup, duplication; LOF, loss of function.
2. Development of the Heart
The heart develops during the early stages of embryogenesis. The cardiac precursor fields differentiate and coalesce to create the cardiac crescent. During human gestation, the heart tube grows, undergoes regional transformation, and emerges as a nearly fully formed organ by 8 weeks. This process involves a linear and then looped structure. During heart development, differentiating precursor cell clusters interact to generate specialised heart cells with a well-defined three-dimensional architecture within restricted regions.
2.1. A Look at the Lines of Progenitor Cells and Their Morphogenesis
The heart is formed by two fields: the first heart field (FHF) from the lateral plate mesoderm and the second heart field (SHF) from the lateral plate splanchnic mesoderm and the migrating cardiac neural crest. These three cell lineages are major contributors to the formation of heart structures in the III, IV, and VI pharyngeal arches [35,41,42,43][35][41][42][43] (Figure 5). The FHF and SHF, which are symmetrical, merge anteriorly to form the heart crescent, which then coalesces into a linear cardiac tube with ventricular and atrial precursors aligned along the anterior–posterior axis. The heart cells originating from the FHF are involved in the development of the atria and the left ventricle, while the cells from the SHF are involved in the development of the right ventricle and the outflow tract, as well as contributing to both atria. Asymmetric growth results in the ballooning of the external curvature of the cardiac tube. This increases the size of the ventricular chamber [44,45][44][45]. The subsequent outflow tract separation and smooth muscle differentiation occur with the contribution of the cardiac neural crest.
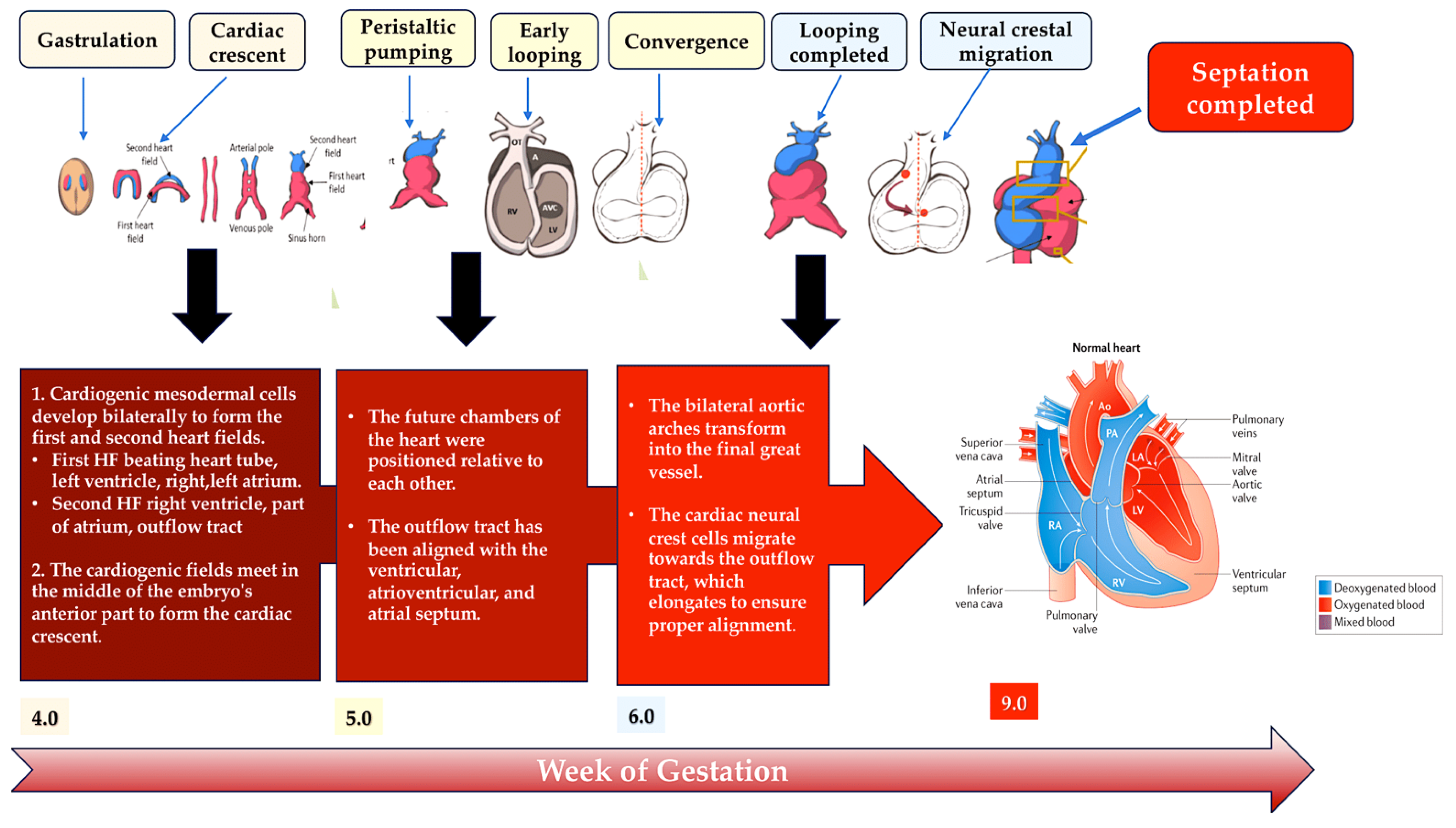
Figure 5. This schematic illustrates the embryonic development of the human heart, including first and second heart field formation, heart tube formation and pumping, looping, neural crest migration, and septation. The process results in a fully developed heart at the end of gestation. Abbreviation: HF, heart field.
2.2. Heart Morphogenesis under Genetic Control
Several important biomolecules play a role in differentiating progenitor cells into cardiac cell lineages and directing cardiac morphogenesis. Genes associated with CHD have identified some of these molecules. The FHF arises from the cardiac mesoderm through signalling, including the bone morphogenetic protein (BMP) and fibroblast growth factor (FGF), which are induced by the adjacent endoderm [46,47,48,49][46][47][48][49]. The expression of key cardiac transcripts required for cardiomyocyte lineage commitment is reduced in experimental models by the deletion of genes encoding BMPs [50]. The expressions of genes such as GATA4, GATA6, NKX2-5, and TBX5 control sarcomere formation and contraction [28,29,30,31,32,33,51][28][29][30][31][32][33][51]. Ventricular cardiomyocyte specification is promoted by the expression of IRX4 in both the FHF and SHF of the cardiac crescent [52]. Factors such as retinoic acid within the mesodermal progenitor fields initiate commitment to an atrial lineage [53,54][53][54]. Further specification leads to cardiomyocytes, which populate the left ventricle (IRX4, HAND1, and MSX1) [55[55][56][57],56,57], the right ventricle (IRX4 and GATA6) [55[55][58],58], and the outflow tract (GATA6, HAND2, ARID3B, and TEA [56,58,59,60][56][58][59][60]. FGF signalling is required for SHF contributions to the outflow tract, allowing for neural crest migration and the induction of tbx1 expression in the SHF in zebrafish [61]. While there are established differentiation pathways, the progenitor fields in cardiac development still maintain plasticity. For instance, removing FHF cells in zebrafish leads to SHF compensation, which regenerates ventricular cardiomyocytes [62]. Additional pathways and gene networks involved in cardiac neural crest differentiation have been discovered. In mice, the normal differentiation of cardiac neural crest cells is prevented in the absence of retinoic acid signalling [63], which can be ameliorated by maternal vitamin A supplementation [64]. This highlights the genotype–environment interactions in CHD. In contrast, a missense variant of GATA6, which is frequently found in patients with CHD and impairs outflow track formation, significantly enhances retinoic acid signalling [58]. FOXC2 expression is essential for cardiac neural crest migration, as it mediates the activation of Sema3c expression in the SHF. TBX1 expression, however, results in the counterbalancing of inhibitory signals [65,66][65][66].
In the atrioventricular canal and outflow tract, the heart contains non-chambered cardiomyocytes, as well as valvular endothelial and smooth muscle cells. Regulatory proteins, such as TBX2, TBX3 [67], BMP2, and BMP4 [68], repress chamber-specific genes to specify the non-chamber myocardium. In order to populate the endocardial cushions, valvular endothelial cells undergo endothelial-to-mesenchymal transition. This process is controlled by signalling cascades. These include vascular endothelial growth factor (VEGF) [69] and transforming growth factor-β (TGFβ) [70]. Investigations into the differentiation of the outflow tract in mouse models have also shown that histone deacetylase 3 (encoded by Hdac3) is necessary for the differentiation of smooth muscle, whereas FGF and sonic hedgehog signalling are needed for the pulmonary arteries and veins [71,72,73][71][72][73]. Through transdifferentiation, cardiomyocytes can also change into cells that are not cardiac chambers. During the development of the mouse heart, a population of cardiomyocytes was found to transdifferentiate into vascular smooth muscle cells of the great arteries [74].
The left–right signalling axis is established in the embryo within the embryonic node. This occurs through signals mediated by motile and sensory cilia, leading to the asymmetric expression of genes that encode downstream regulatory signals.
Among the genes that encode downstream regulatory signals are those that encode the TGFβ family members Nodal, Lefty1, Lefty2, and Zic3 [75,76,77][75][76][77]. These signals are critical for the correct rightward looping of the linear heart tube, which ensures the correct positioning of the atria, ventricles, and outflow tract and gives rise to the sinoatrial node in the right atrium. Knockout mutations in Lefty2, which encodes left–right axis signalling proteins and transcriptional regulators, or the disruption of Sonic hedgehog or activin signalling in the chick embryo [78] can cause defects in cardiac looping, resulting in congenital heart defects in mice [79].
3. New CHD Genes Discovered in Cohort Studies
Analyses of CHD cohorts recruited from single centres or through national and international collaborations are increasingly identifying genes associated with congenital heart disease [22,25,154][22][25][80]. The majority of studies include patients with severe or critical CHD. There is an under-representation of patients with prevalent, simple CHD findings and an absence of patients with confirmed genetic syndromes [155][81]. WES and case–control analyses are commonly used to detect variants. These studies suggest that people with congenital heart disease have similar proportions of total de novo variants to the general population; however, they have significantly higher numbers of rare harmful mutations (allele frequency ≤1 × 10−5 in the general population), including a 9% increase in de novo harmful variants, a 7% higher frequency of dominant inherited variants, and a 1% increase in recessive variants [22,23,24,25][22][23][24][25]. Deleterious mutations are found in an estimated 8–10% of patients with unspecified sporadic CHD [22,25][22][25]. The highest rate of de novo harmful variants is found in patients with syndromic congenital heart disease, while the highest rate of heritable LOF variants is found among patients with isolated CHD [25]. The importance of predefined biological pathways in the pathogenesis of CHD has been supported by cohort studies. The newly identified CHD-associated genes include those involved in the Notch [156][82] and VEGF [156][82] signalling cascades. However, the most significant functional class of these genes is chromatin remodelling [23,36][23][36]. Patients with CHD who have extracardiac anomalies and a neurodevelopmental delay most often have deleterious variants in these genes encoding chromatin remodelling proteins. The broad expression pattern of the genes suggests that organ development uses common epigenetic and transcriptional regulatory pathways. The importance of ciliary function in cardiogenesis is supported by WES data from patients with CHD [157][83]. Deleterious mutations in cilia-related genes are often inherited from a healthy parent, as has been shown in other recessive diseases. In contrast, harmful variations in genes associated with chromatin alterations are more likely to occur de novo, which indicates that the damage caused by even a single mutant allele in this class of genes is significant.-
CHD definitive genes.
-
Genes linked to CHD that have deleterious variants in humans.
References
- Ibrahim, S.; Gaborit, B.; Lenoir, M.; Collod-Beroud, G.; Stefanovic, S. Maternal Pre-Existing Diabetes: A Non-Inherited Risk Factor for Congenital Cardiopathies. Int. J. Mol. Sci. 2023, 24, 16258.
- Dilli, D.; Akduman, H.; Zenciroğlu, A.; Çetinkaya, M.; Okur, N.; Turan, Ö.; Özlü, F.; Çalkavur, Ş.; Demirel, G.; Koksal, N.; et al. Neonatal Outcomes of Critical Congenital Heart Defects: A Multicenter Epidemiological Study of Turkish Neonatal Society: Neonatal Outcomes of CCHD. Pediatr. Cardiol. 2024, 45, 257–271.
- Hossin, M.Z.; de la Cruz, L.F.; McKay, K.A.; Oberlander, T.F.; Sandström, A.; Razaz, N. Association of pre-existing maternal cardiovascular diseases with neurodevelopmental disorders in offspring: A cohort study in Sweden and British Columbia, Canada. Int. J. Epidemiol. 2023, dyad184.
- Bakker, M.K.; Bergman, J.E.H.; Krikov, S.; Amar, E.; Cocchi, G.; Cragan, J.; de Walle, H.E.K.; Gatt, M.; Groisman, B.; Liu, S.; et al. Prenatal diagnosis and prevalence of critical congenital heart defects: An international retrospective cohort study. BMJ Open 2019, 9, e028139.
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900.
- Reller, M.D.; Strickland, M.J.; Riehle-Colarusso, T.; Mahle, W.T.; Correa, A. Prevalence of congenital heart defects in metropolitan Atlanta. J. Pediatr. 2008, 153, 807–813.
- Spector, L.G.; Menk, J.S.; Knight, J.H.; McCracken, C.; Thomas, A.S.; Vinocur, J.M.; Oster, M.E.; St Louis, J.D.; Moller, J.H.; Kochilas, L. Trends in Long-Term Mortality after Congenital Heart Surgery. J. Am. Coll. Cardiol. 2018, 71, 2434–2446.
- Egbe, A.; Lee, S.; Ho, D.; Uppu, S.; Srivastava, S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 2014, 7, 86–91.
- Wang, H.; Lin, X.; Lyu, G.; He, S.; Dong, B.; Yang, Y. Chromosomal abnormalities in fetuses with congenital heart disease: A meta-analysis. Arch. Gynecol. Obstet. 2023, 308, 797–811.
- Diniz, B.L.; Deconte, D.; Gadelha, K.A.; Glaeser, A.B.; Guaraná, B.B.; de Moura, A.Á.; Rosa, R.F.M.; Zen, P.R.G. Congenital Heart Defects and 22q11.2 Deletion Syndrome: A 20-Year Update and New Insights to Aid Clinical Diagnosis. J. Pediatr. Genet. 2023, 12, 113–122.
- Thienpont, B.; Mertens, L.; de Ravel, T.; Eyskens, B.; Boshoff, D.; Maas, N.; Fryns, J.P.; Gewillig, M.; Vermeesch, J.R.; Devriendt, K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007, 28, 2778–2784.
- Wilson, D.I.; Cross, I.E.; Goodship, J.A.; Brown, J.; Scambler, P.J.; Bain, H.H.; Taylor, J.F.; Walsh, K.; Bankier, A.; Burn, J.; et al. A prospective cytogenetic study of 36 cases of DiGeorge syndrome. Am. J. Hum. Genet. 1992, 51, 957–963.
- Sgardioli, I.C.; Vieira, T.P.; Simioni, M.; Monteiro, F.P. Gil-da-Silva-Lopes VL 22q11.2 Deletion Syndrome: Laboratory Diagnosis and TBX1 and FGF8 Mutation Screening. J. Pediatr. Genet. 2015, 4, 17–22.
- Agergaard, P.; Olesen, C.; Østergaard, J.R.; Christiansen, M.; Sørensen, K.M. The prevalence of chromosome 22q11.2 deletions in 2478 children with cardiovascular malformations. A population-based study. Am. J. Med. Genet. A 2012, 158, 498–508.
- Agergaard, P.; Hebert, A.; Sørensen, K.M.; Østergaard, J.R.; Olesen, C. Can clinical assessment detect 22q11.2 deletions in patients with cardiac malformations? A review. Eur. J. Med. Genet. 2011, 54, 3–8.
- Peyvandi, S.; Lupo, P.J.; Garbarini, J.; Woyciechowski, S.; Edman, S.; Emanuel, B.S.; Mitchell, L.E.; Goldmuntz, E. 22q11.2 deletions in patients with conotruncal defects: Data from 1610 consecutive cases. Pediatr. Cardiol. 2013, 34, 1687–1694.
- Dehghan, B.; Sabri, M.R.; Ahmadi, A.; Ghaderian, M.; Mahdavi, C.; Ramezani Nejad, D.; Sattari, M. Identifying the Factors Affecting the Incidence of Congenital Heart Disease Using Support Vector Machine and Particle Swarm Optimization. Adv. Biomed. Res. 2023, 12, 130.
- Pierpont, M.E.; Basson, C.T.; Benson, D.W., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge: A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038.
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 64–72.
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720.
- LaHaye, S.; Corsmeier, D.; Basu, M.; Bowman, J.L.; Fitzgerald-Butt, S.; Zender, G.; Bosse, K.; McBride, K.L.; White, P.; Garg, V. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circ. Cardiovasc. Genet. 2016, 9, 320–329.
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601.
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266.
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438.
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065.
- Shan, W.; Yuanqing, X.; Jing, Z.; Xi, W.; Huifeng, G.; Yi, W. Risk factor analysis for adverse prognosis of the fetal ventricular septal defect (VSD). BMC Pregnancy Childbirth 2023, 23, 683.
- International Society of Ultrasound in Obstetrics & Gynecology. Cardiac screening examination of the fetus: Guidelines for performing the ‘basic’ and ‘extended basic’ cardiac scan. Ultrasound Obstet. Gynecol. 2006, 27, 107–113.
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724.
- Kathiriya, I.S.; Nora, E.P.; Bruneau, B.G. Investigating the transcriptional control of cardiovascular development. Circ. Res. 2015, 116, 700–714.
- Maas, R.G.C.; van den Dolder, F.W.; Yuan, Q.; van der Velden, J.; Wu, S.M.; Sluijter, J.P.G.; Buikema, J.W. Harnessing developmental cues for cardiomyocyte production. Development 2023, 150, dev201483.
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, proliferation, and growth of the mammalian heart. Mol. Ther. 2018, 26, 1599–1609.
- Cui, M.; Wang, Z.; Bassel-Duby, R.; Olson, E.N. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 2018, 145, dev171983.
- Chien, K.R.; Domian, I.J.; Parker, K.K. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science 2008, 322, 1494–1497.
- Jain, R.; Epstein, J.A. Competent for commitment: You’ve got to have heart! Genes Dev. 2018, 32, 4–13.
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268.
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223.
- Hedermann, G.; Hedley, P.L.; Thagaard, I.N.; Krebs, L.; Ekelund, C.K.; Sørensen, T.I.A.; Christiansen, M. Maternal obesity and metabolic disorders associate with congenital heart defects in the offspring: A systematic review. PLoS ONE 2021, 16, e0252343.
- Cavadino, A.; Sandberg, L.; Öhman, I.; Bergvall, T.; Star, K.; Dolk, H.; Loane, M.; Addor, M.C.; Barisic, I.; Cavero-Carbonell, C.; et al. Signal Detection in EUROmediCAT: Identification and Evaluation of Medication-Congenital Anomaly Associations and Use of VigiBase as a Complementary Source of Reference. Drug Saf. 2021, 44, 765–785.
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234.
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Y: Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014.
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease. Semin. Cell Dev. Biol. 2007, 18, 101–110.
- de la Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186.
- Lin, C.-J.; Lin, C.-Y.; Chen, C.-H.; Zhou, B.; Chang, C.-P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299.
- Wu, B.; Wu, B.; Benkaci, S.; Shi, L.; Lu, P.; Park, T.; Morrow, B.E.; Wang, Y.; Zhou, B. Crk and Crkl Are Required in the Endocardial Lineage for Heart Valve Development. J. Am. Heart Assoc. 2023, 12, e029683.
- Christoffels, V.M.; Habets, P.E.; Franco, D.; Campione, M.; de Jong, F.; Lamers, W.H.; Bao, Z.Z.; Palmer, S.; Biben, C.; Harvey, R.P.; et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 2000, 223, 266–278.
- Alsan, B.H.; Schultheiss, T.M. Regulation of avian cardiogenesis by Fgf8 signaling. Development 2002, 129, 1935–1943.
- Astrof, S.; Arriagada, C.; Saijoh, Y.; Francou, A.; Kelly, R.G.; Moon, A. Aberrant differentiation of second heart field mesoderm prefigures cellular defects in the outflow tract in response to loss of FGF8. Dev. Biol. 2023, 499, 10–21.
- Ivanovitch, K.; Soro-Barrio, P.; Chakravarty, P.; Jones, R.A.; Bell, D.M.; Mousavy Gharavy, S.N.; Stamataki, D.; Delile, J.; Smith, J.C.; Briscoe, J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 2021, 19, e3001200.
- Itoh, N.; Ohta, H.; Nakayama, Y.; Konishi, M. Roles of FGF signals in heart development, health, and disease. Front. Cell Dev. Biol. 2016, 4, 110.
- de Pater, E.; Ciampricotti, M.; Priller, F.; Veerkamp, J.; Strate, I.; Smith, K.; Lagendijk, A.K.; Schilling, T.F.; Herzog, W.; Abdelilah-Seyfried, S.; et al. Bmp signaling exerts opposite effects on cardiac differentiation. Circ. Res. 2012, 110, 578–587.
- Targoff, K.L.; Colombo, S.; George, V.; Schell, T.; Kim, S.H.; Solnica-Krezel, L.; Yelon, D. Nkx genes are essential for maintenance of ventricular identity. Development 2013, 140, 4203–4213.
- Nelson, D.O.; Lalit, P.A.; Biermann, M.; Markandeya, Y.S.; Capes, D.L.; Addesso, L.; Patel, G.; Han, T.; John, M.C.; Powers, P.A.; et al. Irx4 Marks a Multipotent, Ventricular-Specific Progenitor Cell. Stem Cells 2016, 34, 2875–2888.
- Goldfracht, I.; Protze, S.; Shiti, A.; Setter, N.; Gruber, A.; Shaheen, N.; Nartiss, Y.; Keller, G.; Gepstein, L. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat. Commun. 2020, 11, 75.
- Giacomelli, E.; Bellin, M.; Orlova, V.V.; Mummery, C.L. Co-Differentiation of Human Pluripotent Stem Cells-Derived Cardiomyocytes and Endothelial Cells from Cardiac Mesoderm Provides a Three-Dimensional Model of Cardiac Microtissue. Curr. Protoc. Hum. Genet. 2017, 95, 21.9.1–21.9.22.
- Cheng, Z.; Wang, J.; Su, D.; Pan, H.; Huang, G.; Li, X.; Li, Z.; Shen, A.; Xie, X.; Wang, B.; et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 2011, 130, 657–662.
- de Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124.
- Chen, Y.H.; Ishii, M.; Sun, J.; Sucov, H.M.; Maxson, R.E. Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 2007, 308, 421–437.
- Sharma, A.; Wasson, L.K.; Willcox, J.A.; Morton, S.U.; Gorham, J.M.; DeLaughter, D.M. Pediatric Cardiac Genomics Consortium. GATA6 mutations in hiPSCs inform mechanisms for maldevelopment of the heart, pancreas, and diaphragm. eLife 2020, 9, e53278.
- Uribe, V.; Badía-Careaga, C.; Casanova, J.C.; Domínguez, J.N.; de la Pompa, J.L.; Sanz-Ezquerro, J.J. Arid3b is essential for second heart field cell deployment and heart patterning. Development 2014, 141, 4168–4181.
- Wang, Z.; Wang, D.Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189.
- Felker, A.; Prummel, K.D.; Merks, A.M.; Mickoleit, M.; Brombacher, E.C.; Huisken, J.; Panáková, D.; Mosimann, C. Continuous addition of progenitors forms the cardiac ventricle in zebrafish. Nat. Commun. 2018, 9, 2001.
- Sánchez-Iranzo, H.; Galardi-Castilla, M.; Minguillón, C.; Sanz-Morejón, A.; González-Rosa, J.M.; Felker, A.; Ernst, A.; Guzmán-Martínez, G.; Mosimann, C.; Mercader, N. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat. Commun. 2018, 9, 428.
- Jiang, X.; Choudhary, B.; Merki, E.; Chien, K.R.; Maxson, R.E.; Sucov, H.M. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev. 2002, 117, 115–122.
- Zhou, C.; Häneke, T.; Rohner, E.; Sohlmér, J.; Kameneva, P.; Artemov, A.; Adameyko, I.; Sahara, M. STRA6 is essential for induction of vascular smooth muscle lineages in human embryonic cardiac outflow tract development. Cardiovasc. Res. 2023, 119, 1202–1217.
- Sanchez, J.; Miyake, R.; Cheng, A.; Liu, T.; Iseki, S.; Kume, T. Conditional inactivation of Foxc1 and Foxc2 in neural crest cells leads to cardiac abnormalities. Genesis 2020, 58, e23364.
- Kodo, K.; Shibata, S.; Miyagawa-Tomita, S.; Ong, S.G.; Takahashi, H.; Kume, T.; Okano, H.; Matsuoka, R.; Yamagishi, H. Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci. Rep. 2017, 7, 6771.
- Greulich, F.; Rudat, C.; Kispert, A. Mechanisms of T-box gene function in the developing heart. Cardiovasc. Res. 2011, 91, 212–222.
- MacGrogan, D.; Nus, M.; de la Pompa, J.L. Notch signaling in cardiac development and disease. Curr. Top. Dev. Biol. 2010, 92, 333–365.
- Stankunas, K.; Ma, G.K.; Kuhnert, F.J.; Kuo, C.J.; Chang, C.P. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev. Biol. 2010, 347, 325–336.
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369.
- Singh, N.; Trivedi, C.M.; Lu, M.; Mullican, S.E.; Lazar, M.A.; Epstein, J.A. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ. Res. 2011, 109, 1240–1249.
- Kelly, R.G.; Papaioannou, V.E. Visualization of outflow tract development in the absence of Tbx1 using an FgF10 enhancer trap transgene. Dev. Dyn. 2007, 236, 821–828.
- Peng, T.; Tian, Y.; Boogerd, C.J.; Lu, M.M.; Kadzik, R.S.; Stewart, K.M.; Evans, S.M.; Morrisey, E. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature 2013, 500, 589–592.
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 2019, 28, 1346–1361.e4.
- Forrest, K.; Barricella, A.C.; Pohar, S.A.; Hinman, A.M.; Amack, J.D. Understanding laterality disorders and the left-right organizer: Insights from zebrafish. Front. Cell Dev. Biol. 2022, 10, 1035513.
- Lopes Floro, K.; Artap, S.T.; Preis, J.I.; Fatkin, D.; Chapman, G.; Furtado, M.B.; Harvey, R.P.; Hamada, H.; Sparrow, D.B.; Dunwoodie, S. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum. Mol. Genet. 2011, 20, 1097–1110.
- Bellchambers, H.M.; Ware, S.M. ZIC3 in Heterotaxy. Adv. Exp. Med. Biol. 2018, 1046, 301–327.
- Levin, M. Left-right asymmetry in vertebrate embryogenesis. Bioessays 1997, 19, 287–296.
- Chang, H.; Zwijsen, A.; Vogel, H.; Huylebroeck, D.; Matzuk, M.M. Smad5 is essential for left-right asymmetry in mice. Dev. Biol. 2000, 219, 71–78.
- Nataf, P.; Guettier, C.; Bourbon, A.; Nappi, F.; Lima, L.; Dorent, R.; Pavie, A.; Gandjbakhch, I. Influence of arterial allograft preparation techniques on chronic vascular rejection: A histological study. Transplant Proc. 1996, 28, 2890–2892.
- Hoang, T.T.; Goldmuntz, E.; Roberts, A.E.; Chung, W.K.; Kline, J.K.; Deanfield, J.E.; Giardini, A.; Aleman, A.; Gelb, B.D.; Mac Neal, M.; et al. The congenital heart disease genetic network study: Cohort description. PLoS ONE 2018, 13, e0191319.
- Preuss, C.; Capredon, M.; Wünnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. MIBAVA Leducq consortium; Samuels ME, Andelfinger G Family based whole exome sequencing reveals the multifaceted role of Notch signaling in congenital heart disease. PLoS Genet. 2016, 12, e1006335.
- Sevim Bayrak, C.; Zhang, P.; Tristani-Firouzi, M.; Gelb, B.D.; Itan, Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020, 12, 9.
- Morton, S.U.; Shimamura, A.; Newburger, P.E.; Opotowsky, A.R.; Quiat, D.; Pereira, A.C.; Jin, S.C.; Gurvitz, M.; Brueckner, M.; Chung, W.K.; et al. Association of damaging variants in genes with increased cancer risk among patients with congenital heart disease. JAMA Cardiol. 2021, 6, 457–462.
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ. Res. 2019, 124, 553–563.
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019, 21, 1001–1007.
- Park, J.E.; Park, J.S.; Jang, S.Y.; Park, S.H.; Kim, J.W.; Ki, C.S.; Kim, D.K. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol. Genet. Genom. Med. 2019, 7, e620.
- Raya, A.; Kawakami, Y.; Rodriguez-Esteban, C.; Buscher, D.; Koth, C.M.; Itoh, T.; Morita, M.; Raya, R.M.; Dubova, I.; Bessa, J.G.; et al. Notch activity induces Nodal expression and mediates the establishment of left-right asymmetry in vertebrate embryos. Genes Dev. 2003, 17, 1213–1218.
- Galvin, K.M.; Donovan, M.J.; Lynch, C.A.; Meyer, R.I.; Paul, R.J.; Lorenz, J.N.; Fairchild-Huntress, V.; Dixon, K.L.; Dunmore, J.H.; Gimbrone, M.A.; et al. A role for Smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000, 24, 171–174.
- McKean, D.M.; Homsy, J.; Wakimoto, H.; Patel, N.; Gorham, J.; DePalma, S.R.; Ware, J.S.; Zaidi, S.; Ma, W.; Patel, N.; et al. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat. Commun. 2016, 7, 12824.
- Brault, V.; Nguyen, T.L.; Flores-Gutiérrez, J.; Iacono, G.; Birling, M.C.; Lalanne, V.; Meziane, H.; Manousopoulou, A.; Pavlovic, G.; Lindner, L.; et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021, 17, e1009777.
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187.
- Reuter, M.S.; Chaturvedi, R.R.; Jobling, R.K.; Pellecchia, G.; Hamdan, O.; Sung, W.W.L.; Nalpathamkalam, T.; Attaluri, P.; Silversides, C.K.; Wald, R.M.; et al. Clinical Genetic Risk Variants Inform a Functional Protein Interaction Network for Tetralogy of Fallot. Circ. Genom. Precis. Med. 2021, 14, e003410.
- Vandeweyer, G.; Helsmoortel, C.; Van Dijck, A.; Vulto-van Silfhout, A.T.; Coe, B.P.; Bernier, R.; Gerdts, J.; Rooms, L.; van den Ende, J.; Bakshi, M.; et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 315–326.
- Ockeloen, C.W.; Willemsen, M.H.; de Munnik, S.; van Bon, B.W.; de Leeuw, N.; Verrips, A.; Kant, S.G.; Jones, E.A.; Brunner, H.G.; van Loon, R.L.; et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur. J. Hum. Genet. 2015, 23, 1176–1185.
- Hamilton, M.J.; Caswell, R.C.; Canham, N.; Cole, T.; Firth, H.V.; Foulds, N.; Heimdal, K.; Hobson, E.; Houge, G.; Joss, S.; et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J. Med. Genet. 2018, 55, 28–38.
- van den Akker, W.M.R.; Brummelman, I.; Martis, L.M.; Timmermans, R.N.; Pfundt, R.; Kleefstra, T.; Willemsen, M.H.; Gerkes, E.H.; Herkert, J.C.; van Essen, A.J.; et al. De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clin. Genet. 2018, 93, 1000–1007.
- Beal, B.; Hayes, I.; McGaughran, J.; Amor, D.J.; Miteff, C.; Jackson, V.; van Reyk, O.; Subramanian, G.; Hildebrand, M.S.; Morgan, A.T.; et al. Expansion of phenotype of DDX3X syndrome: Six new cases. Clin. Dysmorphol. 2019, 28, 169–174.
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291.
More