Immunotherapy is now established as a potent therapeutic paradigm engendering antitumor immune response against a wide range of malignancies and other diseases by modulating the immune system either through the stimulation or suppression of immune components such as CD4+ T cells, CD8+ T cells, B cells, monocytes, macrophages, dendritic cells, and natural killer cells. By targeting several immune checkpoint inhibitors or blockers (e.g., PD-1, PD-L1, PD-L2, CTLA-4, LAG3, and TIM-3) expressed on the surface of immune cells, several monoclonal antibodies and polyclonal antibodies have been developed and already translated clinically. In addition, natural killer cell-based, dendritic cell-based, and CAR T cell therapies have been also shown to be promising and effective immunotherapeutic approaches. In particular, CAR T cell therapy has benefited from advancements in CRISPR-Cas9 genome editing technology, allowing the generation of several modified CAR T cells with enhanced antitumor immunity.
- cancer
- immunogenicity
- immunotherapy
- immune checkpoint
- CAR T cell therapy
- CRISPR-Cas9
1. Introduction
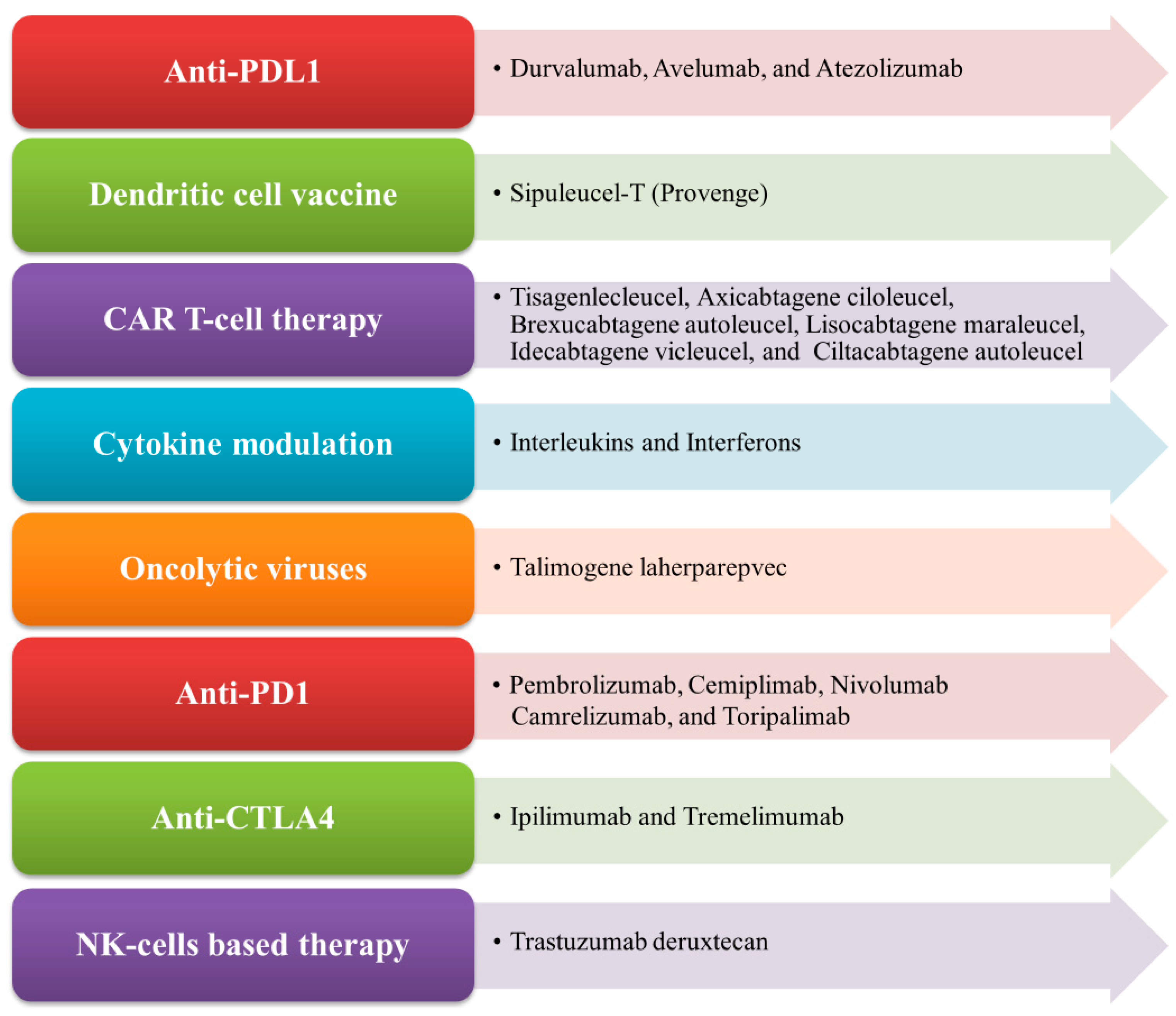
2. Modulation of Tumor Immunogenicity
2.1. Direct Modulation of Tumor Immunogenicity
2.2. Indirect Modulation of Tumor Immunogenicity
2.3. Existing Immunotherapies for the Indirect Modulation of Immunogenicity
2.3.1. Checkpoint Blockade Therapy
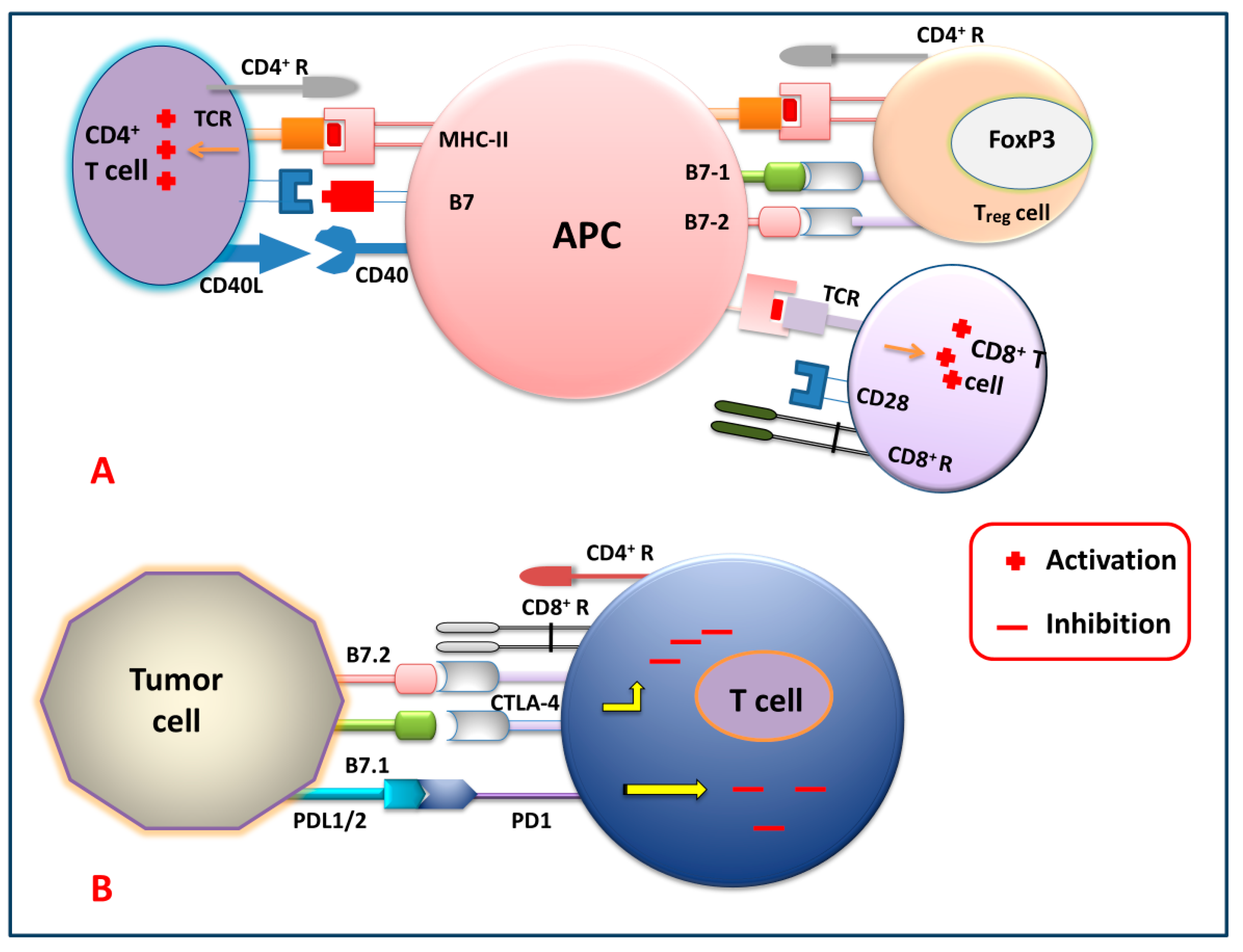
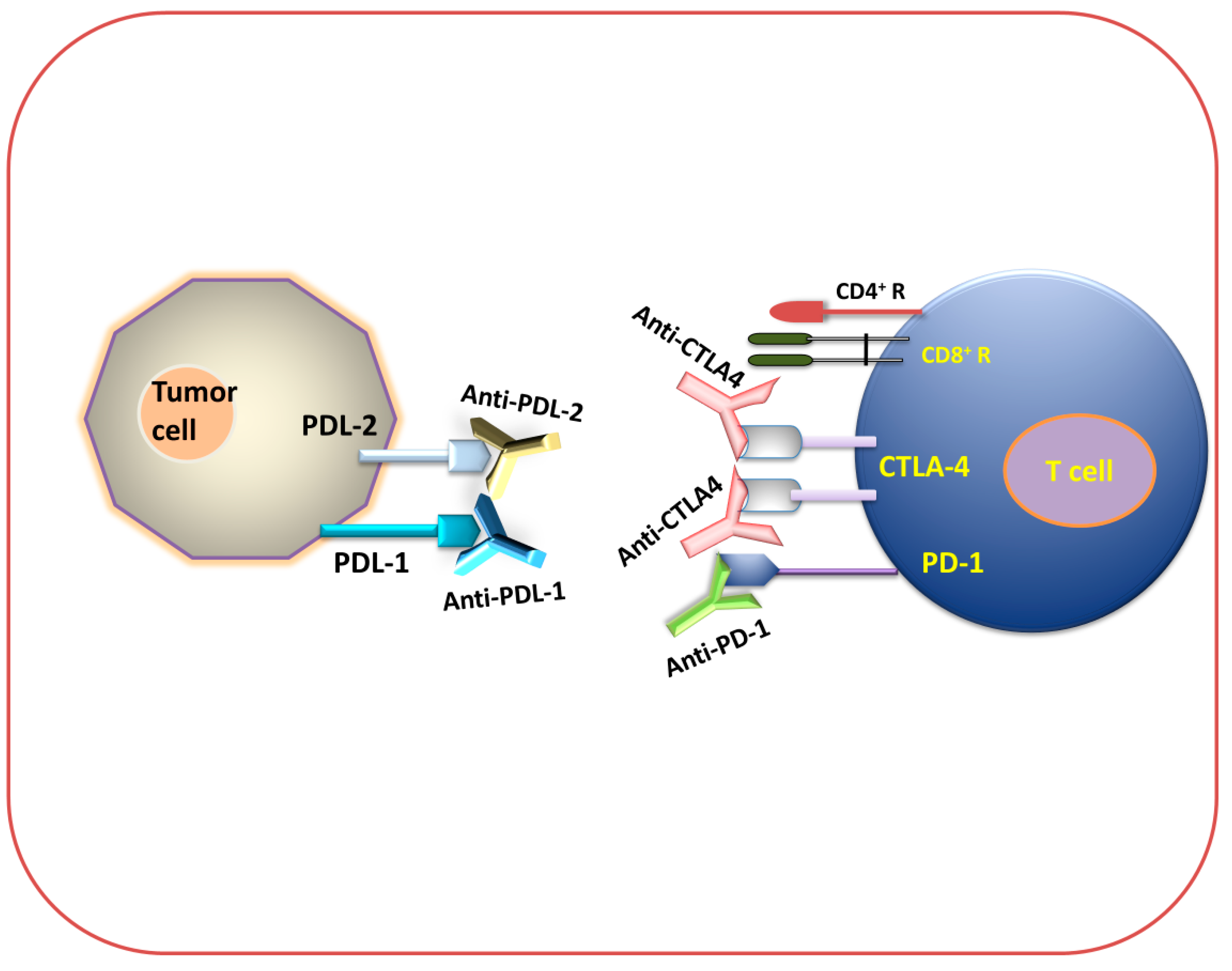
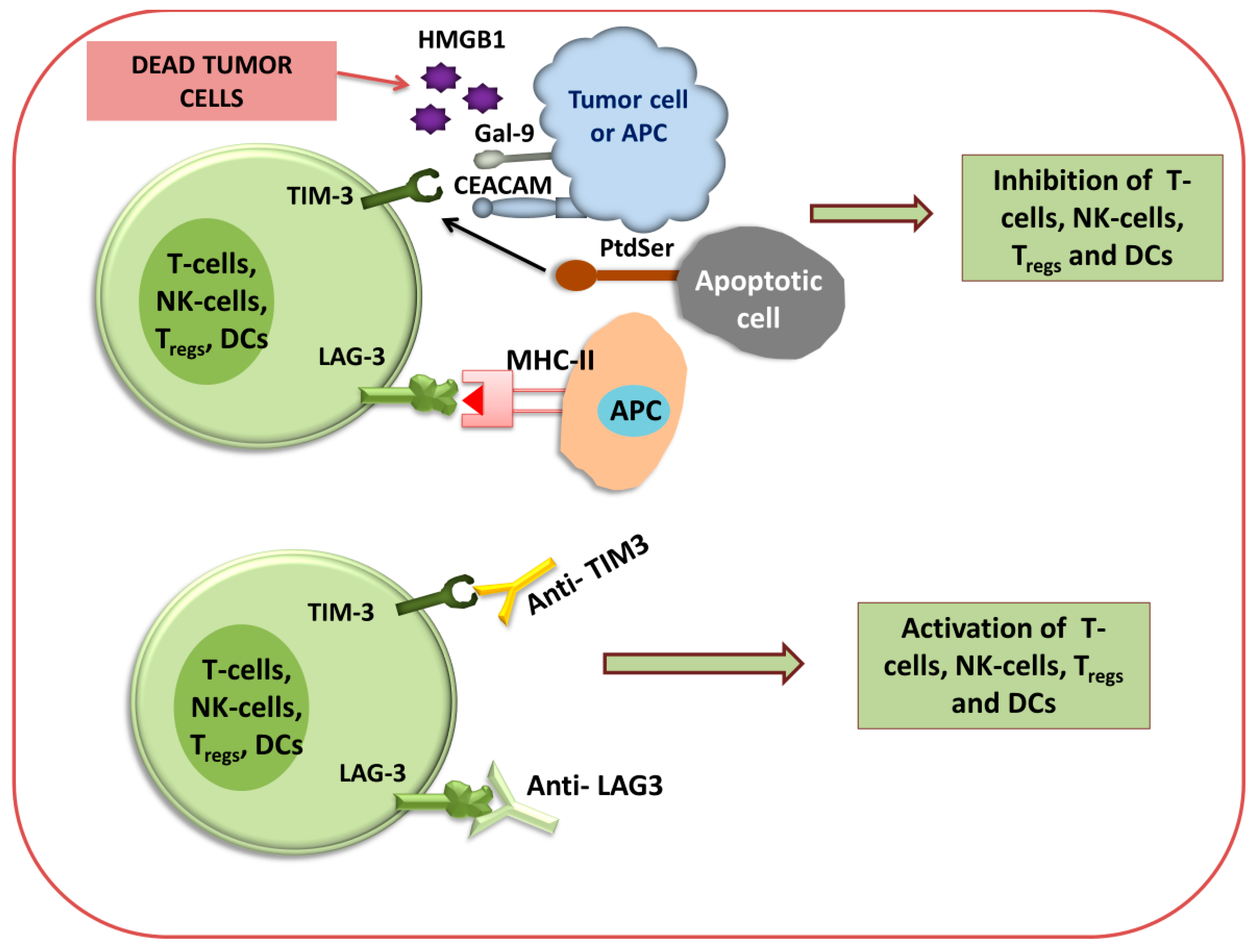
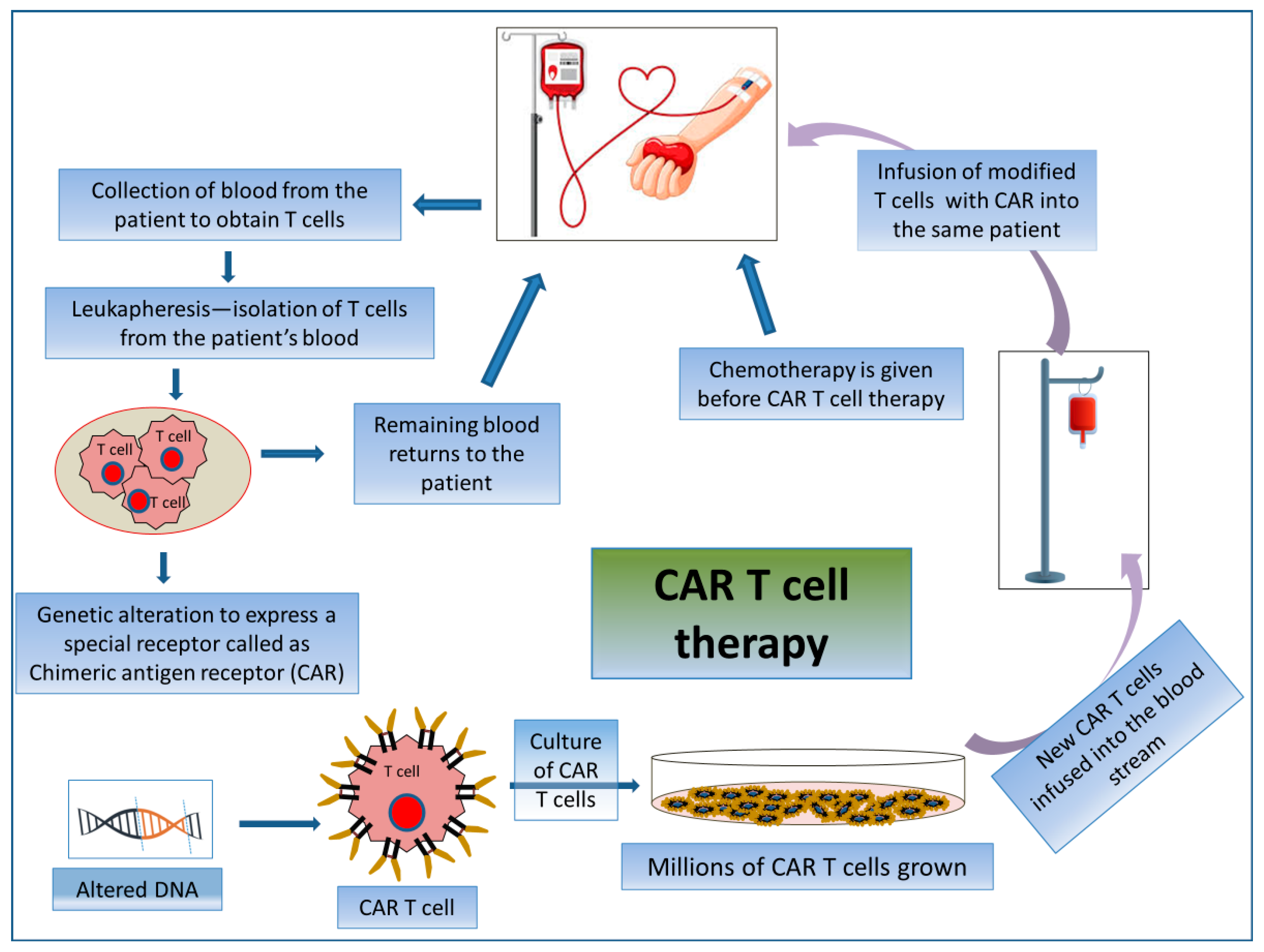
2.3.2. CAR T Cell Therapy
2.3.3. Natural Killer (NK) Cell-Based Immunotherapy (Alternative to CAR T Cell Therapy)
2.3.4. Dendritic Cell Vaccine Therapy (a Cross between a Vaccine and a Cell Therapy)
2.3.5. CRISPR-Cas9-Based Immunotherapy
References
- Esfahani, K.; Elkrief, A.; Calabrese, C.; Lapointe, R.; Hudson, M.; Routy, B.; Miller, W.H., Jr.; Calabrese, L. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 2020, 17, 504–515.
- Wang, Y.; Wang, M.; Wu, H.-X.; Xu, R.-H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829.
- Sharmiladevi, P.; Girigoswami, K.; Haribabu, V.; Girigoswami, A. Nano-enabled theranostics for cancer. Mater. Adv. 2021, 2, 2876–2891.
- Yang, K.; Halima, A.; Chan, T.A. Antigen presentation in cancer—Mechanisms and clinical implications for immunotherapy. Nat. Rev. Clin. Oncol. 2023, 20, 604–623.
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268.
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645.
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer immunotherapy: It’s time to better predict patients’ response. Br. J. Cancer 2021, 125, 927–938.
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360.
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801.
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365.
- Eno, J. Immunotherapy through the years. J. Adv. Pract. Oncol. 2017, 8, 747–753.
- Yun, S.; Vincelette, N.D.; Green, M.R.; Wahner Hendrickson, A.E.; Abraham, I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: A systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016, 5, 1481–1491.
- Bachanova, V.; Miller, J.S. NK cells in therapy of cancer. Crit. Rev. Oncog. 2014, 19, 133–141.
- Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwińska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I.; et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes 2020, 11, 921.
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167.
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498.
- Yalcin, G.D.; Danisik, N.; Baygin, R.C.; Acar, A. Systems Biology and Experimental Model Systems of Cancer. J. Pers. Med. 2020, 10, 180.
- Zhang, J.; Tao, A. Antigenicity, immunogenicity, allergenicity. In Allergy Bioinformatics; Springer Nature B.V.: Dordrecht, The Netherlands, 2015; pp. 175–186.
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519.
- Brode, S.; Cooke, A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit. Rev. Immunol. 2008, 28, 109–126.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Campbell, A.M.; Decker, R.H. Harnessing the Immunomodulatory Effects of Radiation Therapy. Oncology 2018, 32, 370–374.
- Atkins, M.B.; Plimack, E.R.; Puzanov, I.; Fishman, M.N.; McDermott, D.F.; Cho, D.C.; Vaishampayan, U.; George, S.; Olencki, T.E.; Tarazi, J.C.; et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018, 19, 405–415.
- Stanculeanu, D.L.; Daniela, Z.; Lazescu, A.; Bunghez, R.; Anghel, R. Development of new immunotherapy treatments in different cancer types. J. Med. Life 2016, 9, 240–248.
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712.
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311.
- Patel, D.; Bassi, R.; Hooper, A.; Prewett, M.; Hicklin, D.J.; Kang, X. Anti-epidermal growth factor receptor monoclonal antibody cetuximab inhibits EGFR/HER-2 heterodimerization and activation. Int. J. Oncol. 2009, 34, 25–32.
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826.
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127.
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718.
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448.
- García Rubiño, M.E.; Carrillo, E.; Ruiz Alcalá, G.; Domínguez-Martín, A.; Marchal, J.A.; Boulaiz, H. Phenformin as an anticancer agent: Challenges and prospects. Int. J. Mol. Sci. 2019, 20, 3316.
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer Res. 2015, 136, E534–E544.
- Vara-Ciruelos, D.; Dandapani, M.; Russell, F.M.; Grzes, K.M.; Atrih, A.; Foretz, M.; Viollet, B.; Lamont, D.J.; Cantrell, D.A.; Hardie, D.G. Phenformin, but not metformin, delays development of T cell acute lymphoblastic leukemia/lymphoma via cell-autonomous AMPK activation. Cell Rep. 2019, 27, 690–698.
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; di Magliano, M.P.; Swanson, K.D.; Zheng, B. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J. Investig. Dermatol. 2017, 137, 1740–1748.
- Divyapriya, C.; Kannan, A.; Raghavan, V. Expression of CD4, CD8 Biomarkers in Invasive Carcinoma of Breast with Clinicopathological Correlation. J. Pharm. Res. Int. 2021, 33, 65–73.
- Baras, A.S.; Drake, C.; Liu, J.J.; Gandhi, N.; Kates, M.; Hoque, M.O.; Meeker, A.; Hahn, N.; Taube, J.M.; Schoenberg, M.P.; et al. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 2016, 5, e1134412.
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell. Biol. 2018, 96, 21–33.
- Fumet, J.D.; Richard, C.; Ledys, F.; Klopfenstein, Q.; Joubert, P.; Routy, B.; Truntzer, C.; Gagné, A.; Hamel, M.A.; Figueiredo Guimaraes, C.; et al. Prognostic and predictive role of CD8 and PD-L1 determination in lung tumor tissue of patients under anti-PD-1 therapy. Br. J. Cancer 2018, 119, 950–960.
- Huang, L.; Guo, Y.; Liu, S.; Wang, H.; Zhu, J.; Ou, L.; Xu, X. Targeting regulatory T cells for immunotherapy in melanoma. Mol. Biomed. 2021, 2, 11.
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J Med. 2016, 375, 1823–1833.
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920.
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128.
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104.
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 2018, 33, 843–852.
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028.
- Omar, H.A.; Tolba, M.F. Tackling molecular targets beyond PD-1/PD-L1: Novel approaches to boost patients’ response to cancer immunotherapy. Crit. Rev. Oncol. Hematol. 2019, 135, 21–29.
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422.
- Pan, R.Y.; Chung, W.H.; Chu, M.T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 4325874.
- Chong, C.; Coukos, G.; Bassani-Sternberg, M. Identification of tumor antigens with immunopeptidomics. Nat. Biotechnol. 2022, 40, 175–188.
- Thomas, C.; Tampé, R. MHC I assembly and peptide editing—Chaperones, clients, and molecular plasticity in immunity. Curr. Opin. Immunol. 2021, 70, 48–56.
- Liu, C.C.; Steen, C.B.; Newman, A.M. Computational approaches for characterizing the tumor immune microenvironment. Immunology 2019, 158, 70–84.
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392.
- Dersh, D.; Hollý, J.; Yewdell, J.W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128.
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516.
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520.
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11, eaax7918.
- Kim, T.K.; Herbst, R.S.; Chen, L. Defining and understanding adaptive resistance in cancer immunotherapy. Trends Immunol. 2018, 39, 624–631.
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13.
- Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133.
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Webber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144.
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454.
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Savoia, P.; Astrua, C.; Fava, P. Ipilimumab (Anti-Ctla-4 Mab) in the treatment of metastatic melanoma: Effectiveness and toxicity management. Hum. Vaccin. Immunother. 2016, 12, 1092–1101.
- Tartari, F.; Santoni, M.; Burattini, L.; Mazzanti, P.; Onofri, A.; Berardi, R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat. Rev. 2016, 48, 20–24.
- Fumet, J.D.; Isambert, N.; Hervieu, A.; Zanetta, S.; Guion, J.F.; Hennequin, A.; Rederstorff, E.; Bertaut, A.; Ghiringhelli, F. Phase Ib/II trial evaluating the safety, tolerability and immunological activity of durvalumab (MEDI4736) (anti-PD-L1) plus tremelimumab (anti-CTLA-4) combined with FOLFOX in patients with metastatic colorectal cancer. ESMO Open 2018, 3, e000375.
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering car-t cells. Biomark. Res. 2017, 5, 1–6.
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393.
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73.
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626.
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080.
- Rosado-Sánchez, I.; Herrero-Fernández, I.; Genebat, M.; Del Romero, J.; Riera, M.; Podzamczer, D.; Olalla, J.; Vidal, F.; Muñoz-Fernández, M.; Leal, M.; et al. HIV-infected subjects with Poor CD4 T-cell recovery despite effective therapy express high levels of OX40 and α4β7 on CD4 T-cells prior therapy initiation. Front. Immunol. 2018, 9, 1673.
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010, 70, 6725–6734.
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90.
- Li, Y.; Ming, Y.; Fu, R.; Li, C.; Wu, Y.; Jiang, T.; Li, Z.; Ni, R.; Li, L.; Su, H.; et al. The pathogenesis, diagnosis, prevention, and treatment of CAR-T cell therapy-related adverse reactions. Front. Pharmacol. 2022, 13, 950923.
- Chu, J.; Gao, F.; Yan, M.; Zhao, S.; Yan, Z.; Shi, B.; Liu, Y. Natural killer cells: A promising immunotherapy for cancer. J. Transl. Med. 2022, 20, 240.
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100.
- Liu, S.; Galat, V.; Galat4, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7.
- Rees, R.C. MHC restricted and non-restricted killer lymphocytes. Blood Rev. 1990, 4, 204–210.
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326.
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297.
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533.
- Oberschmidt, O.; Kloess, S.; Koehl, U. Redirected primary human chimeric antigen receptor natural killer cells as an “off-the-shelf immunotherapy” for improvement in cancer treatment. Front. Immunol. 2017, 8, 654.
- Khawar, M.B.; Sun, H. CAR-NK Cells: From Natural Basis to Design for Kill. Front. Immunol. 2021, 12, 707542.
- Jong, A.Y.; Wu, C.H.; Li, J.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C. Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J. Extracell. Vesicles 2017, 6, 1294368.
- Mandó, P.; Rivero, S.G.; Rizzo, M.M.; Pinkasz, M.; Levy, E.M. Targeting ADCC: A different approach to HER2 breast cancer in the immunotherapy era. Breast 2021, 60, 15–25.
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3.
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor dendritic cell-based vaccines: Lessons from 20 years of clinical trials and future perspectives. Transl. Res. 2016, 168, 74–95.
- Huber, A.; Dammeijer, F.; Aerts, J.G.J.V.; Vroman, H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for in vitro Antigen Loading of Different DC Subsets? Front. Immunol. 2018, 9, 2804.
- Wimmers, F.; Schreibelt, G.; Sköld, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm shift in dendritic cell-based immunotherapy: From in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front. Immunol. 2014, 5, 165.
- Okamoto, M.; Kobayashi, M.; Yonemitsu, Y.; Koido, S.; Homma, S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 133–138.
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95.
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22.
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48.
- Melief, C.J.; Kast, W.M. Cytotoxic T lymphocyte therapy of cancer and tumor escape mechanisms. Semin. Cancer Biol. 1991, 2, 347–354.
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056.
- Zhang, N.; Bevan, M.J. CD8+ T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168.
- Sutherland, S.I.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307.
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharmacol. Ther. 2011, 36, 197–202.
- Ou, X.; Ma, Q.; Yin, W.; Ma, X.; He, Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front. Cell Dev. Biol. 2021, 9, 674467.
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2062.
- Xia, A.L.; He, Q.F.; Wang, J.C.; Zhu, J.; Sha, Y.Q.; Sun, B.; Lu, X.J. Applications and advances of CRISPR-Cas9 in cancer immunotherapy. J. Med. Genet. 2019, 56, 4–9.
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011.
- Künkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379.
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017, 23, 2255–2266.