Members of the epidermal growth factor receptor (EGFR) family of tyrosine kinase receptors are major regulators of cellular proliferation, differentiation, and survival. In humans, abnormal activation of EGFR is associated with the development and progression of many cancer types, which makes it an attractive target for molecular-guided therapy. Two classes of EGFR-targeted cancer therapeutics include monoclonal antibodies (mAbs), which bind to the extracellular domain of EGFR, and tyrosine kinase inhibitors (TKIs), which mostly target the intracellular part of EGFR and inhibit its activity in molecular signaling. While EGFR-specific mAbs and three generations of TKIs have demonstrated clinical efficacy in various settings, molecular evolution of tumors leads to apparent and sometimes inevitable resistance to current therapeutics, which highlights the need for deeper research in this field.
- epidermal growth factor receptor (EGFR)
- tyrosine kinase inhibitors (TKIs)
- monoclonal antibodies (mAbs)
1. EGF Receptor Protein Family
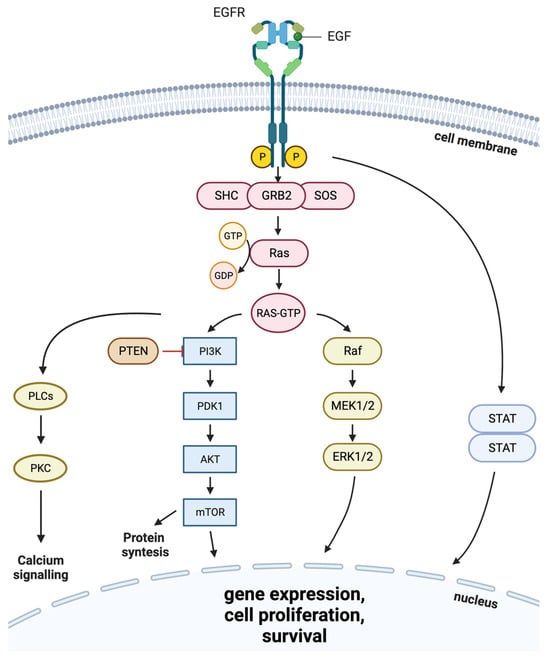
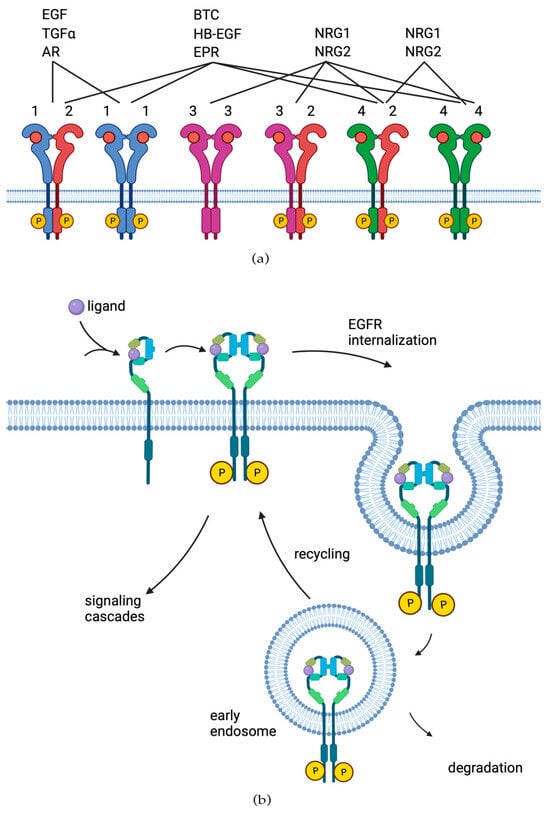
2. EGFR Role in Cancer
Mutations and cases of overexpression of EGFR are especially frequently found in carcinomas and glioblastomas, tumors of epithelial and glial origin, respectively [30,31][27][28]. Worldwide, carcinomas are the most common type of cancer [32][29]. Overexpression of EGFR has been reported and implicated in the pathogenesis of many human malignancies, including head and neck [33][30], lung [34][31], breast [35][32], pancreatic [36][33], and colon cancer [37][34]. The EGFR-positive status of the tumor often correlates with poor prognosis and outcome, as it is beneficial for cancer cell proliferation [7,38][7][35]. EGFR overexpression was also shown to be associated with melanoma progression and promoted invasiveness and metastasis in this tumor type [39][36]. Mutations in the tyrosine kinase domain of EGFR were found in the majority of tumors that exhibited a positive response to treatment with EGFR-specific TKIs (Figure 3a in green) [43][37]. In some reports, the frequency of EGFR-activating mutations has strong ethnical specificity and varies by region, being as high as 46% in Asia versus only 8% in the Americas [44][38]. The two most common mutations of EGFR in NSCLC represent about 85–90% of all EGFR mutations [45][39]. The first one is a deletion of EGFR exon 19 (del747–750), which eliminates the leucine-arginine-glutamate-alanine motif in the tyrosine kinase domain of EGFR (LREA deletion), and the second one (L858R) is a thymine-to-guanine transversion, which results in the replacement of leucine with arginine in exon 21 codon 858 [46,47][40][41]. The third most frequent type of EGFR mutations in NSCLC is exon 20 insertions (ex20ins), which constitute 9% [48][42]–12% [49][43] of all EGFR mutations. In contrast to the other above mentioned mutations, Ex20ins is associated with poor response to treatment with TKIs [50][44]. It results in in-frame insertions, usually concentrated within or following the C-helix that dictates the activation status of EGFR [51][45]. In glioblastoma, the most frequently (~30%) occurring EGFR mutation is EGFRΔIII (EGFR variant III), which results from the in-frame deletion of 801 base pairs spanning exons 2–7 of the coding sequence, resulting in ligand-independent activation of EGFR tyrosine kinase activity [52,53,54][46][47][48].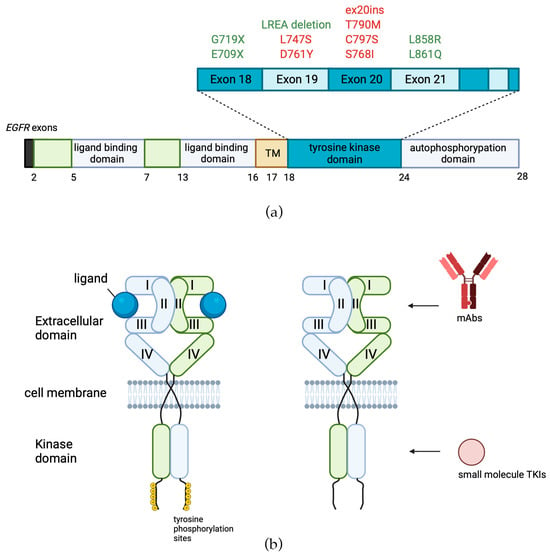
3. EGFR-Targeted Therapies
Since EGFR is frequently overexpressed and/or mutated in multiple cancer types, it has prompted the development of a number of specific targeted therapeutics. Currently, there are two classes of EGFR-specific cancer drugs: monoclonal antibodies (mAbs), which bind to the extracellular domain of the transmembrane receptor and block its dimerization, and small-molecule tyrosine kinase inhibitors (TKIs), which bind to the adenosine triphosphate (ATP) binding site [67][55] (Figure 3b, Table 1). In turn, TKIs can be classified according to the mechanism of binding with the receptor tyrosine kinase domain: type I (binding with ATP site in mainly active conformation), type II (binding with ATP site plus back pocket, DFG(Asp855-Gly857)-out, in inactive conformation), type I½ (binding to a DFG-in, in inactive conformation), type III inhibitors binding to allosteric sites, and type IV inhibitors which generally form covalent adducts with their target protein [68,69][56][57]. EGFR-targeted drugs are currently widespread, globally approved, and are used worldwide for hundreds of thousands of patients per year.| Tyrosine Kinase Inhibitors | |||||
|---|---|---|---|---|---|
| Drug | Tumor Type | Therapeutic Indication | Molecular Target | Inhibitor Type | Molecular Markers of Efficiency |
|
|
First Generation | ||||
| Gefitinib | Advanced or metastatic NSCLC | First-line therapy for NSCLC carrying EGFR-activating mutations | EGFR: ATP-binding site | I | Activating mutations of EGFR: Exon 19 deletions; L858R |
| Erlotinib | Advanced or metastatic NSCLC, pancreatic cancer | First-line therapy for NSCLC carrying EGFR-activating mutations With gemcitabine: first-line treatment option for patients with locally advanced and metastatic pancreatic carcinoma |
EGFR: ATP-binding site | I | Activating mutations of EGFR: Exon 19 deletions; L858R |
| Lapatinib | Metastatic breast cancer | With capecitabine: the treatment of HER2-positive MBC in patients who have previously received therapy (anthracycline, a taxane, trastuzumab) With letrozole: the treatment of postmenopausal women with hormone receptor positive MBC that overexpresses the HER2 receptor for whom hormonal therapy is indicated |
ATP-binding site of EGFR and HER2 | I½ | HER2-positive status of tumor |
|
|
Second Generation | ||||
| Afatinib | Metastatic NSCLC | First-line therapy for metastatic NSCLC carrying EGFR-activating mutations | ATP-binding site of EGFR, HER2, and HER4 | IV | Activating mutations of EGFR: Exon 19 deletions; L858R |
| Neratinib | Breast cancer | Extended adjuvant treatment of patients with early stage HER2-positive breast cancer, to follow adjuvant trastuzumab based therapy With capecitabine: the treatment of patients with advanced or metastatic HER2-positive BC who have received two or more prior anti-HER2 based regimens in the metastatic setting |
ATP-binding site of EGFR, HER2, and HER4 | IV | HER2-positive status of tumor |
| Dacomitinib | Metastatic NSCLC | First-line therapy for metastatic NSCLC carrying EGFR-activating mutations | ATP-binding site of EGFR, HER2, and HER4 | IV | Activating mutations of EGFR: Exon 19 deletions; L858R |
|
|
Third Generation | ||||
| Osimertinib | Advanced or metastatic NSCLC | Adjuvant and first-line therapy for metastatic NSCLC carrying EGFR-activating mutations The treatment of adult patients with metastatic EGFR T790M mutation-positive NSCLC, whose disease has progressed on or after EGFR TKI therapy |
ATP-binding site of the EGFR | IV | Activating mutations of EGFR: Exon 19 deletions; L858R The secondary T790M resistance mutation |
| Almonertinib | Advanced NSCLC | Adjuvant therapy for advanced NSCLC patients with T790M-mutant EGFR who had developed resistance to first- and second-generation EGFR TKIs like gefitinib and afatinib | ATP-binding site of the EGFR | IV | Activating mutations of EGFR: Exon 19 deletions; L858R The secondary T790M resistance mutation |
| Lazertinib | Advanced NSCLC | Treatment of locally advanced or metastatic NSCLC carrying EGFR T790M mutation | ATP-binding site of the EGFR | IV | Activating mutations of EGFR: Exon 19 deletions; L858R The secondary T790M resistance mutation |
| Furmonertinib | Locally advanced or metastatic NSCLC | Treatment of locally advanced or metastatic EGFR T790M+ NSCLC that developed after progression on treatment with first-generation EGFR TKIs | ATP-binding site of the EGFR | The secondary T790M resistance mutation | |
| Monoclonal Antibodies | |||||
| Drug | Tumor Type | Therapeutic Indication | Molecular Target | Molecular Markers of Efficiency | |
| Cetuximab | Advanced or metastatic SCCHN, metastatic CRC | With radiation therapy: treatment of locally or regionally advanced SCCHN With platinum-based therapy with fluorouracil: metastatic SCCHN Metastatic SCCHN progressing after platinum-based therapy With FOLFIRI: first-line treatment of KRASwt EGFR-overexpressing mCRC With irinotecan in patients who are refractory to irinotecan-based chemotherapy: treatment of KRASwt EGFR-overexpressing mCRC; as a single-agent in patients who have failed oxaliplatin-and irinotecan-based chemotherapy or who are intolerant to irinotecan |
The binding site in domain III of EGFR | KRAS wild-type status of EGFR-overexpressing tumor | |
| Panitumumab | Metastatic CRC | Single agent treatment of metastatic CRC with disease progression on or following fluoropyrimidine, oxaliplatin, and irinotecan chemotherapy regimens | The binding site in domain III of EGFR | RAS wild-type status of EGFR-overexpressing tumor | |
| Necitumumab | Metastatic NSCLC | With gemcitabine and cisplatin: first-line treatment of patients with metastatic NSCLC | The binding site in domain III of EGFR | EGFR-overexpressing status of tumor | |
4. First Generation of EGFR-Targeted Drugs
The function of tyrosine kinase enzymes (including those of the ERBB/HER receptor family) is transferring γ-phosphate of an ATP molecule to the tyrosine residue of the substrate, thus initiating signal transmission to further downstream components [70][58]. Thus, targeting the tyrosine kinase activity of EGFR may abrogate its signal transducer capacity inside the cell. -Gefitinib, or ZD1839 (Iressa; Astra-Zeneca Pharmaceuticals), is an oral anilinoquinazolone with a structure formula presented in Figure 4a. By interacting with several amino acid residues, gefitinib takes up space in the ATP-binding site. The first nitrogen of the quinazoline ring creates a hydrogen bond with Met793 in the hinge region and interacts hydrophobically with Leu718, Val726, Lys745, Met766, Leu788, Thr790, and Leu844 residues [73][59].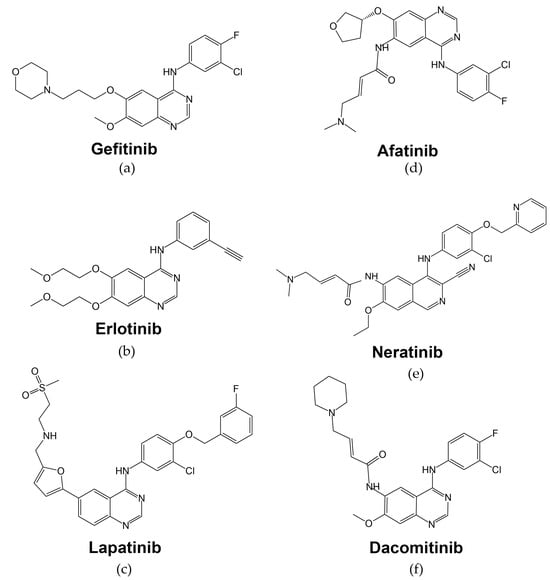
5. Second Generation of EGFR-Targeted Drugs
6. Third Generation of EGFR-Targeted Drugs
First-generation EGFR-targeted low molecular mass therapeutics erlotinib and gefitinib have the disadvantage of being reversible inhibitors, and they are proven to be ineffective against the secondary EGFR mutations, such as the T790M substitution, which has been found in over 50% of EGFR-mutant NSCLC cases with acquired resistance to EGFR inhibitors [159][87]. -Osimertinib (Tagrisso™, AZD9291 AstraZeneca, Figure 5a) is an irreversible orally administered, EGFR-specific TKI with strong selectivity to EGFR-activating mutations as well as the secondary T790M resistance mutation in patients with advanced NSCLC (Figure 5a, Table 1) [160][88]. Osimertinib’s mechanism of action is the formation of a covalent bond to the cysteine-797 residue in the EGFR ATP-binding site [161][89].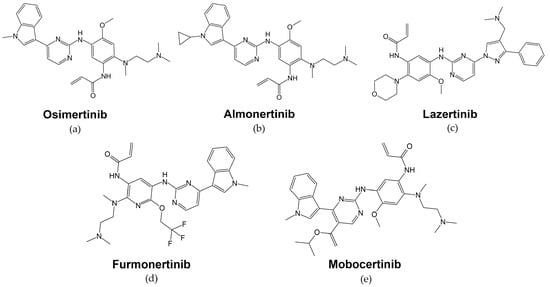
7. Fourth Generation of EGFR-Targeted Drugs
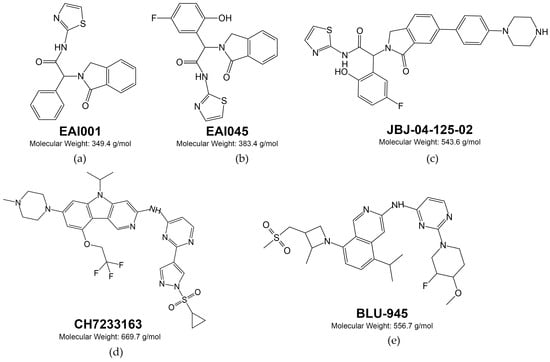
8. EGFR-Specific Therapeutic Monoclonal Antibodies
Soon after the discovery of the EGFR receptor in the 1980s, prof. John Mendelsohn noted that the addition of EGF, the ligand of the EGFR receptor, had a negative effect on the survival of the A431 tumor cell line, which contained large amounts of EGFR. The idea was that it was possible to stop EGFR-overexpressing tumor proliferation through interference with the EGFR signaling [207][107]. Because EGFR permeates the cell membrane, the idea arose that monoclonal antibodies could be an effective therapeutic against tumors with increased expression of this receptor. EGFR-specific mAbs function similarly by disrupting pro-tumor growth and survival signaling through binding to growth factor receptors, thus altering their activation state or preventing ligand binding [208][108]. -Cetuximab (Erbitux, Merck Serono) was the first monoclonal antibody targeting the EGFR receptor, a human–mouse chimeric anti-EGFR mAb with the human IgG1 constant region [212][109]. It exhibits a strong affinity for human EGFR and effectively hinders ligand binding, ultimately resulting in the suppression of receptor phosphorylation and downstream signaling pathways [213][110]. The primary effect of cetuximab binding to EGFR is steric blockage of ligand access to the binding site in domain III of the receptor (Figure 3b, Table 1). It was found that the inhibition of cell growth induced by blocking EGFR activation of cetuximab deals with the induction of cell cycle arrest and apoptosis. Cetuximab induced cell accumulation in the G1 phase and increased the expression levels of cell cycle inhibitors p27KIP1 and p15INK4B in human oral squamous cell carcinoma cell lines [219][111]. A similar effect was observed in SCCHN cell lines: cetuximab treatment decreased cell motility and enhanced cell arrest in the G1 phase; also, the accumulation of p27KIP1 was observed following cetuximab treatment [220][112]. The effectiveness of cetuximab was modulated by the addition of human blood serum of healthy donors to the growth medium in vitro. The addition of 5% human blood serum to cells contributed to a decrease in the antiproliferative activity of cetuximab in the EGFR-overexpressing A431 cell line [222][113], and this effect correlated with a decreased activity of ERK1/2 proteins and repression of cetuximab-induced G1S cell cycle transition arrest. The expression of 75% differently expressed genes, obtained by RNA sequencing, restores to the no-drug level when human serum is added along with cetuximab. The analysis of molecular pathways revealed that the addition of human serum reactivated MAPK signaling pathways inhibited by cetuximab alone [90][64]. Cetuximab has been approved by the European Medicines Agency and the FDA for the use in patients with locally advanced SCCHN and in combination with irinotecan for the treatment of mCRC. In the US, cetuximab has also been approved as monotherapy for patients with recurrent or metastatic SCCHN and in patients with mCRC who cannot tolerate irinotecan-based regimens [229,230][114][115] -Panitumumab (Vectibix, Amgen, Inc., ABX-EGF) is a human monoclonal antibody specifically targeted at EGFR. It has the same presumed mechanism of action as cetuximab, i.e., binding to extracellular domain III of the EGFR molecule and preventing it from activating through interaction with ligands (Table 1) [233][116]. However, panitumumab has a higher affinity for binding with EGFR than cetuximab [234][117]. -Necitumumab (Portrazza, IMC-11F8) is another human monoclonal antibody against EGFR with the same mechanism of action (Table 1) [240][118]. The drug effectively inhibited the growth of tumor cell lines of epidermal, pancreatic, and colorectal origins with EGFR overexpression in vitro [241][119]. Additionally, necitumumab has significant antitumor activity in various human xenograft tumor models and can enhance the antitumor effects of irinotecan and oxaliplatin in colorectal cancer models [242][120].References
- Mitsudomi, T.; Yatabe, Y. Epidermal Growth Factor Receptor in Relation to Tumor Development: EGFR Gene and Cancer: EGFR and Cancer. FEBS J. 2010, 277, 301–308.
- Holbro, T.; Hynes, N.E. ErbB Receptors: Directing Key Signaling Networks throughout Life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217.
- Liu, S.; Geng, R.; Lin, E.; Zhao, P.; Chen, Y. ERBB1/2/3 Expression, Prognosis, and Immune Infiltration in Cutaneous Melanoma. Front. Genet. 2021, 12, 602160.
- Arienti, C.; Pignatta, S.; Tesei, A. Epidermal Growth Factor Receptor Family and Its Role in Gastric Cancer. Front. Oncol. 2019, 9, 1308.
- Roskoski, R. The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 2014, 79, 34–74.
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB Signalling Network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137.
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52.
- Byrne, P.O.; Hristova, K.; Leahy, D.J. EGFR Forms Ligand-Independent Oligomers That Are Distinct from the Active State. J. Biol. Chem. 2020, 295, 13353–13362.
- Mudumbi, K.C.; Burns, E.A.; Schodt, D.J.; Petrova, Z.O.; Kiyatkin, A.; Kim, L.W.; Mangiacapre, E.M.; Ortiz-Caraveo, I.; Ortiz, H.R.; Hu, C.; et al. Distinct Interactions Stabilize EGFR Dimers and Higher-Order Oligomers in Cell Membranes. bioRxiv 2023.
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR Oligomerization Organizes Kinase-Active Dimers into Competent Signalling Platforms. Nat. Commun. 2016, 7, 13307.
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787.
- Singh, B.; Coffey, R.J. From Wavy Hair to Naked Proteins: The Role of Transforming Growth Factor Alpha in Health and Disease. Semin. Cell Dev. Biol. 2014, 28, 12–21.
- Schneider, M.R.; Yarden, Y. Structure and Function of Epigen, the Last EGFR Ligand. Semin. Cell Dev. Biol. 2014, 28, 57–61.
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41.
- Dunbar, A.J.; Goddard, C. Structure-Function and Biological Role of Betacellulin. Int. J. Biochem. Cell Biol. 2000, 32, 805–815.
- Muraoka-Cook, R.S.; Sandahl, M.; Hunter, D.; Miraglia, L.; Earp, H.S. Prolactin and ErbB4/HER4 Signaling Interact via Janus Kinase 2 to Induce Mammary Epithelial Cell Gene Expression Differentiation. Mol. Endocrinol. 2008, 22, 2307–2321.
- Sato, K.; Nakamura, T.; Mizuguchi, M.; Miura, K.; Tada, M.; Aizawa, T.; Gomi, T.; Miyamoto, K.; Kawano, K. Solution Structure of Epiregulin and the Effect of Its C-Terminal Domain for Receptor Binding Affinity. FEBS Lett. 2003, 553, 232–238.
- Ozaki, M. Neuregulins and the Shaping of Synapses. Neuroscientist 2001, 7, 146–154.
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A Novel Neural Tissue-Enriched Protein That Binds and Activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567.
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A Novel Growth Factor That Acts through the ErbB-4 Receptor Tyrosine Kinase. Oncogene 1999, 18, 2681–2689.
- Henriksen, L.; Grandal, M.V.; Knudsen, S.L.J.; van Deurs, B.; Grøvdal, L.M. Internalization Mechanisms of the Epidermal Growth Factor Receptor after Activation with Different Ligands. PLoS ONE 2013, 8, e58148.
- Leblanc, J.A.; Sugiyama, M.G.; Antonescu, C.N.; Brown, A.I. Quantitative Modeling of EGF Receptor Ligand Discrimination via Internalization Proofreading. Phys. Biol. 2023, 20, 056008.
- Landgraf, R. HER2 Therapy. HER2 (ERBB2): Functional Diversity from Structurally Conserved Building Blocks. Breast Cancer Res. 2007, 9, 202.
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A Hierarchical Network of Interreceptor Interactions Determines Signal Transduction by Neu Differentiation Factor/Neuregulin and Epidermal Growth Factor. Mol. Cell. Biol. 1996, 16, 5276–5287.
- Jones, R.B.; Gordus, A.; Krall, J.A.; MacBeath, G. A Quantitative Protein Interaction Network for the ErbB Receptors Using Protein Microarrays. Nature 2006, 439, 168–174.
- Jura, N.; Shan, Y.; Cao, X.; Shaw, D.E.; Kuriyan, J. Structural Analysis of the Catalytically Inactive Kinase Domain of the Human EGF Receptor 3. Proc. Natl. Acad. Sci. USA 2009, 106, 21608–21613.
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The Epidermal Growth Factor Receptor Variant III (EGFRvIII): Where Wild Things Are Altered. FEBS J. 2013, 280, 5350–5370.
- Ohgaki, H.; Kleihues, P. Genetic Alterations and Signaling Pathways in the Evolution of Gliomas. Cancer Sci. 2009, 100, 2235–2241.
- Hinck, L.; Näthke, I. Changes in Cell and Tissue Organization in Cancer of the Breast and Colon. Curr. Opin. Cell Biol. 2014, 26, 87–95.
- Kalyankrishna, S.; Grandis, J.R. Epidermal Growth Factor Receptor Biology in Head and Neck Cancer. J. Clin. Oncol. 2006, 24, 2666–2672.
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal Growth Factor Receptor (EGFR) in Lung Cancer: An Overview and Update. J. Thorac. Dis. 2010, 2, 48–51.
- Al-Kuraya, K.; Schraml, P.; Torhorst, J.; Tapia, C.; Zaharieva, B.; Novotny, H.; Spichtin, H.; Maurer, R.; Mirlacher, M.; Köchli, O.; et al. Prognostic Relevance of Gene Amplifications and Coamplifications in Breast Cancer. Cancer Res. 2004, 64, 8534–8540.
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers 2011, 3, 1513–1526.
- Pabla, B.; Bissonnette, M.; Konda, V.J. Colon Cancer and the Epidermal Growth Factor Receptor: Current Treatment Paradigms, the Importance of Diet, and the Role of Chemoprevention. World J. Clin. Oncol. 2015, 6, 133–141.
- Herbst, R.S. Review of Epidermal Growth Factor Receptor Biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26.
- Pastwińska, J.; Karaś, K.; Karwaciak, I.; Ratajewski, M. Targeting EGFR in Melanoma—The Sea of Possibilities to Overcome Drug Resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188754.
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139.
- Graham, R.P.; Treece, A.L.; Lindeman, N.I.; Vasalos, P.; Shan, M.; Jennings, L.J.; Rimm, D.L. Worldwide Frequency of Commonly Detected EGFR Mutations. Arch. Pathol. Lab. Med. 2018, 142, 163–167.
- Castañeda-González, J.P.; Chaves, J.J.; Parra-Medina, R. Multiple Mutations in the EGFR Gene in Lung Cancer: A Systematic Review. Transl. Lung Cancer Res. 2022, 11, 2148–2163.
- Choi, Y.W.; Jeon, S.Y.; Jeong, G.S.; Lee, H.W.; Jeong, S.H.; Kang, S.Y.; Park, J.S.; Choi, J.-H.; Koh, Y.W.; Han, J.H.; et al. EGFR Exon 19 Deletion Is Associated with Favorable Overall Survival After First-Line Gefitinib Therapy in Advanced Non-Small Cell Lung Cancer Patients. Am. J. Clin. Oncol. 2018, 41, 385–390.
- Yermekova, S.; Orazgaliyeva, M.; Goncharova, T.; Rakhimbekova, F.; Dushimova, Z.; Vasilieva, T. Mutational Damages in Malignant Lung Tumors. Asian Pac. J. Cancer Prev. 2023, 24, 709–716.
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural History and Molecular Characteristics of Lung Cancers Harboring EGFR Exon 20 Insertions. J. Thorac. Oncol. 2013, 8, 179–184.
- Burnett, H.; Emich, H.; Carroll, C.; Stapleton, N.; Mahadevia, P.; Li, T. Epidemiological and Clinical Burden of EGFR Exon 20 Insertion in Advanced Non-Small Cell Lung Cancer: A Systematic Literature Review. PLoS ONE 2021, 16, e0247620.
- Wang, F.; Li, C.; Wu, Q.; Lu, H. EGFR Exon 20 Insertion Mutations in Non-Small Cell Lung Cancer. Transl. Cancer Res. 2020, 9, 2982–2991.
- Vyse, S.; Huang, P.H. Targeting EGFR Exon 20 Insertion Mutations in Non-Small Cell Lung Cancer. Signal Transduct. Target. Ther. 2019, 4, 5.
- Rutkowska, A.; Stoczyńska-Fidelus, E.; Janik, K.; Włodarczyk, A.; Rieske, P. EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Zheng, Q.; Han, L.; Dong, Y.; Tian, J.; Huang, W.; Liu, Z.; Jia, X.; Jiang, T.; Zhang, J.; Li, X.; et al. JAK2/STAT3 Targeted Therapy Suppresses Tumor Invasion via Disruption of the EGFRvIII/JAK2/STAT3 Axis and Associated Focal Adhesion in EGFRvIII-Expressing Glioblastoma. Neuro-Oncology 2014, 16, 1229–1243.
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat. Rev. Cancer 2007, 7, 169–181.
- Tsigelny, I.F.; Wheler, J.J.; Greenberg, J.P.; Kouznetsova, V.L.; Stewart, D.J.; Bazhenova, L.; Kurzrock, R. Molecular Determinants of Drug-Specific Sensitivity for Epidermal Growth Factor Receptor (EGFR) Exon 19 and 20 Mutants in Non-Small Cell Lung Cancer. Oncotarget 2015, 6, 6029–6039.
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227.
- Garima, G.; Thanvi, S.; Singh, A.; Verma, V. Epidermal Growth Factor Receptor Variant III Mutation, an Emerging Molecular Marker in Glioblastoma Multiforme Patients: A Single Institution Study on the Indian Population. Cureus 2022, 14, e26412.
- Li, Y.; Zhang, H.-B.; Chen, X.; Yang, X.; Ye, Y.; Bekaii-Saab, T.; Zheng, Y.; Zhang, Y. A Rare EGFR-SEPT14 Fusion in a Patient with Colorectal Adenocarcinoma Responding to Erlotinib. Oncologist 2020, 25, 203–207.
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851.
- Gharwan, H.; Groninger, H. Kinase Inhibitors and Monoclonal Antibodies in Oncology: Clinical Implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227.
- Roskoski, R. Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol. Res. 2019, 139, 395–411.
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48.
- Meng, Y.; Pond, M.P.; Roux, B. Tyrosine Kinase Activation and Conformational Flexibility: Lessons from Src-Family Tyrosine Kinases. Acc. Chem. Res. 2017, 50, 1193–1201.
- Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819.
- Suenaga, M.; Yamaguchi, A.; Soda, H.; Orihara, K.; Tokito, Y.; Sakaki, Y.; Umehara, M.; Terashi, K.; Kawamata, N.; Oka, M.; et al. Antiproliferative Effects of Gefitinib Are Associated with Suppression of E2F-1 Expression and Telomerase Activity. Anticancer Res. 2006, 26, 3387–3391.
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa) Tablets. Oncologist 2003, 8, 303–306.
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva) Tablets. Oncologist 2005, 10, 461–466.
- Shan, F.; Shao, Z.; Jiang, S.; Cheng, Z. Erlotinib Induces the Human Non-Small-Cell Lung Cancer Cells Apoptosis via Activating ROS-Dependent JNK Pathways. Cancer Med. 2016, 5, 3166–3175.
- Kamashev, D.; Shaban, N.; Lebedev, T.; Prassolov, V.; Suntsova, M.; Raevskiy, M.; Gaifullin, N.; Sekacheva, M.; Garazha, A.; Poddubskaya, E.; et al. Human Blood Serum Can Diminish EGFR-Targeted Inhibition of Squamous Carcinoma Cell Growth through Reactivation of MAPK and EGFR Pathways. Cells 2023, 12, 2022.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final Overall Survival Results from a Randomised, Phase III Study of Erlotinib versus Chemotherapy as First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883.
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib): Relationships among Protein Conformation, Inhibitor off-Rate, and Receptor Activity in Tumor Cells. Cancer Res. 2004, 64, 6652–6659.
- Ongko, J.; Setiawan, J.V.; Feronytha, A.G.; Juliana, A.; Effraim, A.; Wahjudi, M.; Antonius, Y. In-Silico Screening of Inhibitor on Protein Epidermal Growth Factor Receptor (EGFR). IOP Conf. Ser. Earth Environ. Sci. 2022, 1041, 012075.
- Tevaarwerk, A.J.; Kolesar, J.M. Lapatinib: A Small-Molecule Inhibitor of Epidermal Growth Factor Receptor and Human Epidermal Growth Factor Receptor-2 Tyrosine Kinases Used in the Treatment of Breast Cancer. Clin. Ther. 2009, 31 Pt 2, 2332–2348.
- Liu, L.; Zhong, L.; Zhao, Y.; Chen, M.; Yao, S.; Li, L.; Xiao, C.; Shan, Z.; Gan, L.; Xu, T.; et al. Effects of Lapatinib on Cell Proliferation and Apoptosis in NB4 Cells. Oncol. Lett. 2018, 15, 235–242.
- Cameron, D.; Casey, M.; Oliva, C.; Newstat, B.; Imwalle, B.; Geyer, C.E. Lapatinib plus Capecitabine in Women with HER-2-Positive Advanced Breast Cancer: Final Survival Analysis of a Phase III Randomized Trial. Oncologist 2010, 15, 924–934.
- Dungo, R.T.; Keating, G.M. Afatinib: First Global Approval. Drugs 2013, 73, 1503–1515.
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017, 56, 235–250.
- Banno, E.; Togashi, Y.; Kobayashi, Y.; Hayashi, H.; Mitsudomi, T.; Nishio, K. Afatinib Is Especially Effective against Non-Small Cell Lung Cancer Carrying an EGFR Exon 19 Deletion. Anticancer Res. 2015, 35, 2005–2008.
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an Irreversible EGFR/HER2 Inhibitor Highly Effective in Preclinical Lung Cancer Models. Oncogene 2008, 27, 4702–4711.
- Young, N.R.; Soneru, C.; Liu, J.; Grushko, T.A.; Hardeman, A.; Olopade, O.I.; Baum, A.; Solca, F.; Cohen, E.E.W. Afatinib Efficacy against Squamous Cell Carcinoma of the Head and Neck Cell Lines In Vitro and In Vivo. Target. Oncol. 2015, 10, 501–508.
- Schuler, M.; Wu, Y.-L.; Hirsh, V.; O’Byrne, K.; Yamamoto, N.; Mok, T.; Popat, S.; Sequist, L.V.; Massey, D.; Zazulina, V.; et al. First-Line Afatinib versus Chemotherapy in Patients with Non-Small Cell Lung Cancer and Common Epidermal Growth Factor Receptor Gene Mutations and Brain Metastases. J. Thorac. Oncol. 2016, 11, 380–390.
- Awada, A.; Dirix, L.; Manso Sanchez, L.; Xu, B.; Luu, T.; Diéras, V.; Hershman, D.L.; Agrapart, V.; Ananthakrishnan, R.; Staroslawska, E. Safety and Efficacy of Neratinib (HKI-272) plus Vinorelbine in the Treatment of Patients with ErbB2-Positive Metastatic Breast Cancer Pretreated with Anti-HER2 Therapy. Ann. Oncol. 2013, 24, 109–116.
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor Activity of HKI-272, an Orally Active, Irreversible Inhibitor of the HER-2 Tyrosine Kinase. Cancer Res. 2004, 64, 3958–3965.
- Wissner, A.; Mansour, T.S. The Development of HKI-272 and Related Compounds for the Treatment of Cancer. Arch. Pharm. 2008, 341, 465–477.
- Conlon, N.T.; Kooijman, J.J.; van Gerwen, S.J.C.; Mulder, W.R.; Zaman, G.J.R.; Diala, I.; Eli, L.D.; Lalani, A.S.; Crown, J.; Collins, D.M. Comparative Analysis of Drug Response and Gene Profiling of HER2-Targeted Tyrosine Kinase Inhibitors. Br. J. Cancer 2021, 124, 1249–1259.
- Asami, K.; Atagi, S. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors for Non-Small Cell Lung Cancer. World J. Clin. Oncol. 2014, 5, 646–659.
- Nagano, T.; Tachihara, M.; Nishimura, Y. Dacomitinib, a Second-Generation Irreversible Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor (EGFR-TKI) to Treat Non-Small Cell Lung Cancer. Drugs Today 2019, 55, 231–236.
- Garuti, L.; Roberti, M.; Bottegoni, G. Irreversible Protein Kinase Inhibitors. Curr. Med. Chem. 2011, 18, 2981–2994.
- Duggirala, K.B.; Lee, Y.; Lee, K. Chronicles of EGFR Tyrosine Kinase Inhibitors: Targeting EGFR C797S Containing Triple Mutations. Biomol. Ther. 2022, 30, 19–27.
- Engelman, J.A.; Zejnullahu, K.; Gale, C.-M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an Irreversible Pan-ERBB Inhibitor, Is Effective in Lung Cancer Models with EGFR and ERBB2 Mutations That Are Resistant to Gefitinib. Cancer Res. 2007, 67, 11924–11932.
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patients with Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180.
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061.
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121, 725–737.
- Yang, J.C.-H.; Camidge, D.R.; Yang, C.-T.; Zhou, J.; Guo, R.; Chiu, C.-H.; Chang, G.-C.; Shiah, H.-S.; Chen, Y.; Wang, C.-C.; et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients with EGFR-Mutated Advanced NSCLC: A Multicenter, Open-Label, Phase 1 Trial. J. Thorac. Oncol. 2020, 15, 1907–1918.
- Yun, J.; Hong, M.H.; Kim, S.-Y.; Park, C.-W.; Kim, S.; Yun, M.R.; Kang, H.N.; Pyo, K.-H.; Lee, S.S.; Koh, J.S.; et al. YH25448, an Irreversible EGFR-TKI with Potent Intracranial Activity in EGFR Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 2575–2587.
- Zhang, S.S.; Ou, S.-H.I. Spotlight on Furmonertinib (Alflutinib, AST2818). The Swiss Army Knife (Del19, L858R, T790M, Exon 20 Insertions, “Uncommon-G719X, S768I, L861Q”) Among the Third-Generation EGFR TKIs? Lung Cancer 2022, 13, 67–73.
- Vasconcelos, P.E.N.S.; Kobayashi, I.S.; Kobayashi, S.S.; Costa, D.B. Preclinical Characterization of Mobocertinib Highlights the Putative Therapeutic Window of This Novel EGFR Inhibitor to EGFR Exon 20 Insertion Mutations. JTO Clin. Res. Rep. 2021, 2, 100105.
- Arnold, A.; Ganti, A.K. Clinical Utility of Mobocertinib in the Treatment of NSCLC—Patient Selection and Reported Outcomes. OncoTargets Ther. 2023, 16, 559–569.
- Wu, L.; Ke, L.; Zhang, Z.; Yu, J.; Meng, X. Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 602762.
- Papadimitrakopoulou, V.A.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Bulusu, K.C.; Laus, G.; et al. Analysis of Resistance Mechanisms to Osimertinib in Patients with EGFR T790M Advanced NSCLC from the AURA3 Study. Ann. Oncol. 2018, 29, viii741.
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534.
- Papini, F.; Sundaresan, J.; Leonetti, A.; Tiseo, M.; Rolfo, C.; Peters, G.J.; Giovannetti, E. Hype or Hope—Can Combination Therapies with Third-Generation EGFR-TKIs Help Overcome Acquired Resistance and Improve Outcomes in EGFR-Mutant Advanced/Metastatic NSCLC? Crit. Rev. Oncol. Hematol. 2021, 166, 103454.
- Zhao, P.; Yao, M.-Y.; Zhu, S.-J.; Chen, J.-Y.; Yun, C.-H. Crystal Structure of EGFR T790M/C797S/V948R in Complex with EAI045. Biochem. Biophys. Res. Commun. 2018, 502, 332–337.
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132.
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943.
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297.
- Conti, C.; Campbell, J.; Woessner, R.; Guo, J.; Timsit, Y.; Iliou, M.; Wardwell, S.; Davis, A.; Chicklas, S.; Hsieh, J.; et al. Abstract 1262: BLU-701 Is a Highly Potent, Brain-Penetrant and WT-Sparing next-Generation EGFR TKI for the Treatment of Sensitizing (Ex19del, L858R) and C797S Resistance Mutations in Metastatic NSCLC. Cancer Res. 2021, 81, 1262.
- Tavera, L.; Schalm, S.; Campbell, J.; Guo, J.; Medendorp, C.; Chen, M.; Albayya, F.; Dineen, T.; Zhang, Z.; Iliou, M.; et al. Abstract 3328: Antitumor Activity of BLU-945 and BLU-701 as Single Agents and in Combination in EGFR L858R-Driven Models of NSCLC. Cancer Res. 2022, 82, 3328.
- Spira, A.I.; Spigel, D.R.; Camidge, D.R.; De Langen, A.; Kim, T.M.; Goto, K.; Elamin, Y.Y.; Shum, E.; Reckamp, K.L.; Rotow, J.K.; et al. A Phase 1/2 Study of the Highly Selective EGFR Inhibitor, BLU-701, in Patients with EGFR-Mutant Non–Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2022, 40, TPS9142.
- Yun, M.R.; Yu, M.R.; Duggirala, K.B.; Lee, K.; Jo, A.; Seah, E.; Kim, C.; Cho, B.C. MA07.08 JIN-A02, a Highly Effective 4th Generation EGFR-TKI, Targeting EGFR C797S Triple Mutation in NSCLC. J. Thorac. Oncol. 2022, 17, S69–S70.
- Kawamoto, T.; Sato, J.D.; Le, A.; Polikoff, J.; Sato, G.H.; Mendelsohn, J. Growth Stimulation of A431 Cells by Epidermal Growth Factor: Identification of High-Affinity Receptors for Epidermal Growth Factor by an Anti-Receptor Monoclonal Antibody. Proc. Natl. Acad. Sci. USA 1983, 80, 1337–1341.
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34.
- Galizia, G.; Lieto, E.; De Vita, F.; Orditura, M.; Castellano, P.; Troiani, T.; Imperatore, V.; Ciardiello, F. Cetuximab, a Chimeric Human Mouse Anti-Epidermal Growth Factor Receptor Monoclonal Antibody, in the Treatment of Human Colorectal Cancer. Oncogene 2007, 26, 3654–3660.
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological Efficacy of a Chimeric Antibody to the Epidermal Growth Factor Receptor in a Human Tumor Xenograft Model. Clin. Cancer Res. 1995, 1, 1311–1318.
- Kiyota, A.; Shintani, S.; Mihara, M.; Nakahara, Y.; Ueyama, Y.; Matsumura, T.; Tachikawa, T.; Wong, D.T.W. Anti-Epidermal Growth Factor Receptor Monoclonal Antibody 225 Upregulates P27KIP1 and P15INK4B and Induces G1 Arrest in Oral Squamous Carcinoma Cell Lines. Oncology 2002, 63, 92–98.
- Okuyama, K.; Suzuki, K.; Naruse, T.; Tsuchihashi, H.; Yanamoto, S.; Kaida, A.; Miura, M.; Umeda, M.; Yamashita, S. Prolonged Cetuximab Treatment Promotes p27Kip1-Mediated G1 Arrest and Autophagy in Head and Neck Squamous Cell Carcinoma. Sci. Rep. 2021, 11, 5259.
- Kamashev, D.; Sorokin, M.; Kochergina, I.; Drobyshev, A.; Vladimirova, U.; Zolotovskaia, M.; Vorotnikov, I.; Shaban, N.; Raevskiy, M.; Kuzmin, D.; et al. Human Blood Serum Can Donor-Specifically Antagonize Effects of EGFR-Targeted Drugs on Squamous Carcinoma Cell Growth. Heliyon 2021, 7, e06394.
- Information on Cetuximab (Marketed as Erbitux)|FDA. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/information-cetuximab-marketed-erbitux (accessed on 17 November 2023).
- Blick, S.K.A.; Scott, L.J. Cetuximab: A Review of Its Use in Squamous Cell Carcinoma of the Head and Neck and Metastatic Colorectal Cancer. Drugs 2007, 67, 2585–2607.
- Voigt, M.; Braig, F.; Göthel, M.; Schulte, A.; Lamszus, K.; Bokemeyer, C.; Binder, M. Functional Dissection of the Epidermal Growth Factor Receptor Epitopes Targeted by Panitumumab and Cetuximab. Neoplasia 2012, 14, 1023–1031.
- Kim, G.P.; Grothey, A. Targeting Colorectal Cancer with Human Anti-EGFR Monoclonocal Antibodies: Focus on Panitumumab. Biologics 2008, 2, 223–228.
- Fala, L. Portrazza (Necitumumab), an IgG1 Monoclonal Antibody, FDA Approved for Advanced Squamous Non-Small-Cell Lung Cancer. Am. Health Drug Benefits 2016, 9, 119–122.
- Lu, D.; Zhang, H.; Koo, H.; Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A Fully Human Recombinant IgG-like Bispecific Antibody to Both the Epidermal Growth Factor Receptor and the Insulin-like Growth Factor Receptor for Enhanced Antitumor Activity. J. Biol. Chem. 2005, 280, 19665–19672.
- Kuenen, B.; Witteveen, P.O.; Ruijter, R.; Giaccone, G.; Dontabhaktuni, A.; Fox, F.; Katz, T.; Youssoufian, H.; Zhu, J.; Rowinsky, E.K.; et al. A Phase I Pharmacologic Study of Necitumumab (IMC-11F8), a Fully Human IgG1 Monoclonal Antibody Directed Against EGFR in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2010, 16, 1915–1923.