Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Miquéias Lopes-Pacheco.
The implementation of cystic fibrosis (CF) transmembrane conductance regulator (CFTR) modulator drugs into clinical practice has been attaining remarkable therapeutic outcomes for CF, a life-threatening autosomal recessive genetic disease. However, there is elevated CFTR allelic heterogeneity, and various individuals carrying (ultra)rare CF genotypes remain without any approved modulator therapy. Novel translational model systems based on individuals’ own cells/tissue are now available and can be used to interrogate in vitro CFTR modulator responses and establish correlations of these assessments with clinical features, aiming to provide prediction of therapeutic effectiveness.
- airway cells
- bioassay
- biomarkers
- extracellular vesicles
- inflammation
- microRNA
- precision medicine
- organoids
- theratyping
1. Introduction
Inherited in an autosomal recessive pattern, cystic fibrosis (CF) is a life-threatening progressive disease affecting over 100,000 people worldwide [1,2][1][2]. The disease occurs due to mutations in the gene encoding the CF transmembrane conductance (CFTR) protein [3[3][4][5],4,5], a chloride/bicarbonate channel expressed at the apical plasma membrane (PM) that plays a vital role in regulating fluid and ion movements across several epithelial tissues, including lungs, intestine, pancreas, and sweat glands [1,6][1][6]. However, despite the multiorgan features of the disease, the elevated morbidity and mortality of people with CF (PwCF) are linked to an accelerated decline of lung function due to repeated cycles of airway mucus accumulation, chronic inflammation, and persistent infection that lead to tissue remodeling and respiratory failure [6,7][6][7].
CF diagnosis is based on symptomatology consistent with the disease and laboratory biomarkers that provide evidence of CFTR dysfunction [8,9][8][9]. Most new cases of CF are nowadays identified within newborn screening programs by assessing immunoreactive trypsinogen in bloodspots. To confirm the diagnosis, CFTR (dys)function should be demonstrated by: (i) identification of CFTR pathogenic variants in both alleles by genetic analysis, and (ii) elevated sweat chloride concentration (SCC; >60 mmol·L−1), altered transepithelial nasal potential difference (NPD) and/or intestinal current measurement (ICM) [8,9][8][9]. Although SCC is the standard test for CF diagnosis, the alternative biomarkers are particularly relevant in cases in which clinical signs and symptoms are suggestive of CF, but SCC is intermediate (30–59 mmol·L−1) [8,9][8][9].
Over 2100 variants have been identified in the CFTR gene [10], of which approximately one-third are now classified as CF-causing [11]. The deletion of a phenylalanine at position 508 (p.Phe508del, legacy: F508del) is the most prevalent CF-causing variant, accounting for approximately 70% of all CF alleles [1], while the remaining 30% of CF alleles are represented by an enormous number of CFTR variants and most are (ultra)rare, occurring among few PwCF worldwide [11]. Due to such CFTR allelic heterogeneity, distinct CF phenotypes exist—on average, PwCF with pancreatic insufficient exhibit more severe forms of the disease, while milder phenotypes are usually associated with pancreatic sufficiency [12]. Indeed, these variants cause distinct primary defects, comprising CFTR mRNA and protein biosynthesis, anion transport, and/or PM turnover. Therefore, they have been separated into CFTR variant classes, which are characterized by alterations in (I) expression, (II) folding and trafficking, (III) gating, (IV) conductance, (V) abundance, and (VI) PM stability [13,14][13][14]. Overall, CFTR variants in classes I and II are associated with a minimal (or null) function, while a residual (or some) function is usually observed in those variants in classes IV–VI. This grouping offers the advantage that CFTR variants with similar defects might be tackled by similar therapeutic strategies—i.e., theratyping [15].
Over the last two decades, precision (or personalized) medicines targeting the fundamental cause of CF have been developed with tremendous accomplishments attained [14,16][14][16]. Indeed, four CFTR modulator drugs are now approved for clinical use: the correctors lumacaftor (LUMA, VX-809), tezacaftor (TEZA, VX-661), and elexacaftor (ELX, VX-445), which retrieve CFTR folding and trafficking to the PM, and the potentiator ivacaftor (IVA, VX-770), which enhances CFTR channel open probability. These drugs—particularly the ‘highly effective’ ones—have provided impressive clinical benefits, representing thus a new dawn for PwCF with eligible genotypes [17,18,19,20,21,22,23][17][18][19][20][21][22][23]. However, there is a significant number of PwCF carrying (ultra)rare variants for whom no modulator therapy has been approved [11,16][11][16].
 While cells heterologously expressing CF-causing variants remain very useful for CFTR studies, primary cell models can provide a more sensitive and reliable prediction of therapeutic responses for several reasons: (i) The last express CFTR in the native genomic context, while cell lines frequently use CFTR cDNA (a copy of the mature mRNA, which lacks the introns); therefore, cells lines may not recapitulate certain cellular mechanisms, including nonsense-mediated decay or splicing effects. For instance, p.Gly970Arg (legacy G970R) was thought to be a CFTR gating variant based on cDNA expression findings [30,31][25][26]; however, analysis of cells from PwCF carrying this variant revealed that it actually causes a splicing defect [32,33][27][28]. (ii) The cellular background has a marked influence on CFTR processing and function as well as its pharmacological sensitivity. As exemplified by the cases of p.Phe508del [34,35][29][30] and p.Gly1244Glu [36][31], the complexity of cellular processes related to protein biogenesis and folding, as well as its PM trafficking, may not be completely recapitulated in cell lines—particularly those from non-human and/or non-respiratory epithelium origin [29][24]. Likewise, CFTR gene regulation can be impacted by epigenetic factors and these are only taken into account by the assessment in primary cells from each individual [37][32]. (iii) Characterization of single variants can be efficiently accomplished in cell lines; however, two variants indicating to be low responsive (below therapeutic relevant threshold) in separated cell lines can compose a genotype with a good prediction for clinical benefits (if evidenced by assessing responses in this individual’s cells). This is particularly relevant for (ultra)rare CFTR variants that are frequently identified in racial and ethnic minority populations, which are usually excluded from traditional clinical trial designs [38][33].
Moreover, numerous reports have established correlations of the data on primary cell models with clinical features of PwCF (before and after initiating modulator therapy) to provide a translational perspective of therapeutic effects (Table 1). Accordingly, these can serve as potential biomarkers to identify which drug(s) could be the best therapeutics for every individual with CF—i.e., “the right therapy for the right person”.
While cells heterologously expressing CF-causing variants remain very useful for CFTR studies, primary cell models can provide a more sensitive and reliable prediction of therapeutic responses for several reasons: (i) The last express CFTR in the native genomic context, while cell lines frequently use CFTR cDNA (a copy of the mature mRNA, which lacks the introns); therefore, cells lines may not recapitulate certain cellular mechanisms, including nonsense-mediated decay or splicing effects. For instance, p.Gly970Arg (legacy G970R) was thought to be a CFTR gating variant based on cDNA expression findings [30,31][25][26]; however, analysis of cells from PwCF carrying this variant revealed that it actually causes a splicing defect [32,33][27][28]. (ii) The cellular background has a marked influence on CFTR processing and function as well as its pharmacological sensitivity. As exemplified by the cases of p.Phe508del [34,35][29][30] and p.Gly1244Glu [36][31], the complexity of cellular processes related to protein biogenesis and folding, as well as its PM trafficking, may not be completely recapitulated in cell lines—particularly those from non-human and/or non-respiratory epithelium origin [29][24]. Likewise, CFTR gene regulation can be impacted by epigenetic factors and these are only taken into account by the assessment in primary cells from each individual [37][32]. (iii) Characterization of single variants can be efficiently accomplished in cell lines; however, two variants indicating to be low responsive (below therapeutic relevant threshold) in separated cell lines can compose a genotype with a good prediction for clinical benefits (if evidenced by assessing responses in this individual’s cells). This is particularly relevant for (ultra)rare CFTR variants that are frequently identified in racial and ethnic minority populations, which are usually excluded from traditional clinical trial designs [38][33].
Moreover, numerous reports have established correlations of the data on primary cell models with clinical features of PwCF (before and after initiating modulator therapy) to provide a translational perspective of therapeutic effects (Table 1). Accordingly, these can serve as potential biomarkers to identify which drug(s) could be the best therapeutics for every individual with CF—i.e., “the right therapy for the right person”.
2. Laboratory Tools to Predict CFTR Modulator Effectiveness In Vivo
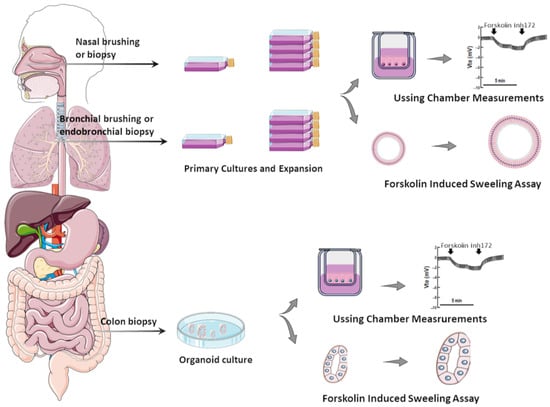
Several in vitro assays have been developed using various model systems to comparatively assess the efficacy of CFTR modulators (individually or combinations thereof) [29][24]. Although heterologous cell lines have been fundamental to enhance the understanding of CFTR biology at genetic, biochemical, and physiological levels, in the context of precision medicine, they can only be used for variant theratyping (i.e., matching single variants to modulators), being unable to predict responses of a determined individual to a specific therapy. Accordingly, alternative translational models have been established by using primary cells from PwCF to inquire about responses at an individual level (Figure 1).Figure 1. Translational human CF model systems for personalized/precision medicine. Airway samples can be collected from the nose or bronchi by brushing or biopsies and gastrointestinal samples can be collected from the colon or rectum by biopsies. These samples are cultured in specific in vitro conditions, expanded, and then seeded on porous membranes to grow in polarized monolayers or in matrigel to form organoids/spheroids. Cell monolayers and organoids can be used to assess CFTR function/rescue through Ussing chamber measurements and through forskolin-induced swelling assays, respectively.
Table 1.
Studies reporting correlations of CFTR modulator-promoted responses in cell models and clinical effects in PwCF.
| Model System | CF Population/Genotypes | CFTR Modulator(s) | Key Findings/Correlations | Ref. |
|---|---|---|---|---|
| HNE cells | PwCF homozygous for p.Phe508del or carrying genotypes leading to minimal or residual CFTR function | LUMA TEZA IVA |
The level of CFTR rescue significantly correlated with the ppFEV1 change at 6 months in 8 PwCF treated with CFTR modulators. | [25][34] |
| Ten PwCF (<19 years) with a wide range of CFTR variants | LUMA IVA |
CFTR modulation produced changes in CFTR function in nasal and bronchial cell cultures. There was a correlation between the residual CFTR function in both cell types from PwCF with SCC between 60 and 90 mmol·L−1. |
[39][35] | |
| Five healthy subjects (they were not genetically tested) and eight PwCF with variants associated with minimal or residual CFTR function | – | CFTR-mediated transepithelial chloride currents measured in vitro through Ussing chamber assay correlated with the SCC of subjects. | [40][36] | |
| Twelve adults with CF with either the p.Gly551Asp or p.ArgR117His | IVA | A strong correlation between changes in SCC and in vitro CFTR activation was observed. Furthermore, a moderate correlation between in vitro CFTR activation and changes in ppFEV1 was reported. | [41][37] | |
| Seven PwCF (≤16 years, clinically stable) with residual CFTR function variants |
IVA | All subjects with decreased SCC in response to IVA treatment also had significant increases in chloride current in HNE cultures with IVA exposure. | [42][38] | |
| Healthy volunteers, PwCF with p.PheF508del/p.Arg117His-7T, p.PheF508del/p.PheF508del, p.PheF508del/p.Met284fs, p.Arg334Trp/c.406-1G>A, and p.Ser18ArgfsX16/p.Ser18ArgfsX16 | LUMA TEZA IVA |
In vitro CFTR chloride measurements correlated to changes in SCC. | [43][39] | |
| Five PwCF: one homozygous for p.Ser737Phe and four compound heterozygous for p.Ser737Phe | TEZA ELX IVA |
In vitro analysis demonstrated different levels of CFTR activity. Some degree of CFTR dysfunction was detected by evaluating chloride secretion in HNE cells derived from compound heterozygous subjects; CFTR activity was improved by IVA alone and even more by treatment with correctors. | [44][40] | |
| Eleven PwCF carrying FDA-eligible variants for ELX/TEZA/IVA and twenty-eight PwCF carrying non-eligible variants | TEZA ELX IVA |
There was a significant relationship between CFTR activity correction and changes in ppFEV1 or SCC. | [45][41] | |
| A 56-year-old male with CF and the p.Phe508del/p.Gln1291His genotype | TEZA ELX IVA |
In HNE cells, p.Phe508del/p.Gln1291His resulted in reduced baseline CFTR activity, and showed minimal response to ELX/TEZA/IVA (individually or in combination), aligning with the individual’s clinical evaluation as a non-responder to these drugs. | [46][42] | |
| Airway organoids | Nine healthy volunteers and three PwCF | LUMA IVA |
Organoids treated with modulators displayed similar effects to clinical response; LUMA/IVA treatment promoted greater responses in organoids from p.PheF508del-homozygous subjects. | [26][43] |
| Six healthy volunteers, nine PwCF homozygous for p.Phe508del, and ten with at least one non-p.Phe508del variant | LUMA IVA |
p.Phe508del/p.Phe508del organoids treated with LUMA/IVA exhibited a positive change in swelling responses; a relationship between organoid response to drug and in vivo clinical response was observed for three subjects treated. | [47][44] | |
| Eighteen PwCF and five healthy volunteers | TEZA ELX IVA |
In vitro responses to CFTR modulators correlated well with clinical measurements. | [48][45] | |
| Five PwCF: p.Phe508del/p.Phe508del, p.Phe508del/p.Gly542X, and F508del/G542X and p.Arg334Trp/p.Arg334Trp | LUMA IVA |
Responses of organoids correlated well with clinical findings. | [49][46] | |
| Intestinal organoids | Twelve healthy volunteers, four CF carriers (WT/p.Phe508del), thirty-five PwCF carrying class II and III variants, and eighteen PwCF carrying class IV and V variants |
LUMA IVA |
Responses of organoids to CFTR modulators correlated with outcome data from clinical trials. | [24][47] |
| Twenty-four PwCF: fifteen carrying at least one p.Ser1251Asn and nine carrying at least one (ultra)rare CFTR variant | LUMA IVA |
Responses to CFTR modulators in organoids correlated with in vivo measurements (SCC and ppFEV1). | [50][48] | |
| Thirty-four children with CF carrying a wide range of CFTR variants | – | Organoid swelling significantly correlated with SCC and ICM. | [51][49] | |
| Thirty-four PwCF with p.Phe508del/p.Phe508del | – | Variability in CFTR residual function appeared to contribute to the clinical heterogeneity and organoid swelling values; responses in organoids correlated with ppFEV1 and BMI. | [52][50] | |
| Ninety-seven PwCF with well-characterized and rare CFTR variants | LUMA IVA |
Measurements of residual CFTR function and rescue by CFTR modulators in organoids correlated with clinical data, namely changes in ppFEV1 and SCC. | [53][51] | |
| A 56-year-old female with CF carrying p.Phe508del/c.3717+5G>T | LUMA TEZA IVA |
No baseline swelling was observed in organoids, suggesting minimal CFTR function; however, limited swelling was detected after LUMA/IVA or TEZA/IVA treatment. | [54][52] | |
| Twenty-one PwCF homozygous for p.Phe508del | LUMA IVA |
No correlations were found between organoid swelling and changes in the in vivo biomarkers, namely SCC and NPD. | [55][53] | |
| A total of 173 PwCF carrying a wide range of CFTR variants | – | Organoid swelling values were associated with long-term ppFEV1 decline and the probability of developing different CF-related comorbidities, namely pancreatic insufficiency, CF-related liver disease, and CF-related diabetes. | [56][54] | |
| Fifteen PwCF homozygous for p.Phe508del, fifteen PwCF carrying p.Phe508del/class I variant, and twenty-two PwCF with rare variant non-eligible for CFTR modulator therapy | TEZA ELX IVA |
Responses to modulators in organoids from p.Phe508del/p.Phe508del or p.Phe508del/class I variant correlated with changes in ppFEV1. In CF organoids with 11 rare genotypes, CFTR function restoration was reported upon ELX/TEZA/IVA treatment. |
[57][55] | |
| A 19-year-old female with CF carrying p.Tyr515X/p.Arg334Trp | LUMA TEZA ELX IVA |
Organoids treated with IVA alone or in combination with correctors demonstrated similar rescue of CFTR-dependent fluid secretion. In vivo measurements demonstrated significant clinical improvements in SCC, ppFEV1, and respiratory symptoms after 7 days of IVA initiation that were sustained for the 9-month follow-up. |
[58][56] | |
| A 6-year-old male with CF carrying p.Phe508del/p.Glu217Gly-Gly509Asp | LUMA TEZA ELX IVA |
The p.Glu217Gly-Gly509Asp complex allele was characterized by a high residual function of the CFTR channel; organoid swelling values demonstrated rescue of CFTR function by all tested modulators. | [59][57] |
Abbreviations: BMI—body mass index; CF—cystic fibrosis; CFTR—CF transmembrane conductance regulator; ELX—elexacaftor; HNE—human nasal epithelial; ICM—intestinal current measurement; IVA—ivacaftor; LUMA—lumacaftor; NPD—nasal potential differences; PwCF—people with CF; ppFEV1—percent predicted forced expiratory volume in 1 s; SCC—sweat chloride concentration; TEZA—tezacaftor.
2.1. Primary Airway Cells Grown in Monolayers
Since the development of the first CFTR modulators, primary human bronchial epithelial (HBE) cells have been considered the gold standard to confirm the efficacy of these drugs in vitro for the subsequent clinical assessment [60,61,62][58][59][60]. These cells can be obtained either from the lungs of individuals undergoing transplant or by bronchial brushing. However, despite the development of well-established protocols to expand and maintain HBE cells to high passage numbers [63][61], bronchoscopy is a considerably invasive procedure that requires sedation and anesthesia. Likewise, the need for explanted lungs limits the availability of these cells, particularly of (ultra)rare CF genotypes. Such limitations were overcome by the adoption of a method of conditional reprogramming of cells [64,65,66][62][63][64] and the usage of cells from the nasal epithelium [39,67][35][65], which have become routinely used by several CF research groups. Nasal epithelial cells can be obtained through minimally invasive procedures, such as nasal brushing or scraping of the lower turbinates [65[63][65],67], which is well tolerated by children and adults with CF and does not require sedation or anesthesia. When cultured under conditional reprogramming conditions, human nasal epithelial (HNE) cells acquire progenitor stem-cell-like features, enabling their expansion with prolonged lifespan and differentiation into various cell types of the respiratory tract [40,65,66,68,69][36][63][64][66][67]. Although epithelial cell populations can be distinct in the upper and lower airways [70[68][69][70],71,72], studies comparing HNE and HBE cells differentiated at the air–liquid interface (ALI) demonstrated that they exhibit similar morpho-functional properties and response to inflammatory cytokines [39,48,73][35][45][71]. HBE and HNE cells from the same individual also demonstrated equivalent CFTR-mediated anion transport in electrophysiological measurements [25,39][34][35]. The analysis of CFTR function in these cells relies primarily on the bioelectric movement of ion transport assessed in micro-Ussing chambers or patch clamps [74,75][72][73]. It is notable that both HBE and HNE cells are highly sensitive to culture conditions [63][61]; therefore, it is imperative to standardize protocols and reference cells to ensure reproducibility among different operators and laboratories. Several reports have indicated that HNE cells can be successfully used as a surrogate for HBE cells in CFTR studies and theratyping [25,39,40][34][35][36]. It is notable that strong correlations were described in the in vitro rescue of CFTR function by modulator drugs in both cell types and in vivo alterations in SCC [39][35]. Data from IVA-promoted CFTR-mediated chloride transport in HNE cells also correlated well with alterations in SCC and ppFEV1 of PwCF carrying either p.Arg117His (legacy: R117H) or p.Gly551Asp (legacy: G551D) [41][37]. Responses of CFTR function to modulator drugs in HNE cultures also demonstrated a good correlation with alterations in SCC, ICM, and lung function (measured as percent predicted forced expiratory volume in one second [ppFEV1]) of PwCF carrying rare genotypes [40,43,44,45,76][36][39][40][41][74] or homozygous for p.Phe508del [25,77][34][75]. Furthermore, modulator-promoted responses in HNE cultures have been assessed to identify non-eligible responders for compassionate use [45][41]. Altogether, these studies indicate that measuring CFTR function in HNE cultures serves as a good predictor of clinical benefits that can be subsequently verified in vivo to enhance the access of CFTR modulator drugs for PwCF carrying common and rare variants.2.2. Airway Organoids/Nasospheroids
Because the primary assessment of CFTR function in HBE and HNE cultures is based on micro-Ussing chamber measurements, which is a low-throughput technique, protocols have been optimized to culture these cells into 3D models [26,47,49,78][43][44][46][76], allowing thus for the assessment of CFTR function in high throughput. Initial studies found that these 3D models can recapitulate various features of the in vivo airway epithelia, including expression of tight junctions, cilia, and mucins [79][77], and assessment of CFTR-mediated fluid secretion on airway organoids enables to discriminate CF and non-CF cultures [26,47][43][44]. Furthermore, CFTR-mediated chloride transport in micro-Ussing chamber measurements of HNE cultures were found to closely correlate with forskolin-induced swelling (FIS) assay of airway organoids. The latter is a microscopy-based functional assay in which CFTR function can be indirectly measured based on fluid movement upon CFTR stimulation by forskolin. When CFTR is activated/rescued, an increase in organoid size/swelling occurs [29,80][24][78]. Airway organoids can be generated in two configurations [26,49][43][46]: (i) With the apical membrane located at the inside due to the presence of a physical matrix (e.g., matrigel) in the culture. In this case, CFTR activation leads to organoid swelling, since fluid secretion occurs from the basal to the apical side. (ii) On the other hand, the omission of a physical matrix in the culture enables the formation of organoids with the apical membrane located outside, and CFTR activation leads thus to organoid shrinking [26,49][43][46]. For the in vitro assessment of CFTR function/rescue, the first configuration and the FIS assay have been the most broadly employed recently [29,49,80][24][46][78]. Upon rescue of CFTR using modulator drugs, responses were demonstrated in organoids from PwCF carrying p.Phe508del in both alleles [26,47][43][44] or a range of rare CFTR variants [48,49][45][46]. Furthermore, CFTR baseline and modulator-rescued responses in airway organoids demonstrated a significant correlation with alterations in SCC and ppFEV1 [48][45]. Despite such progress, further development and refining of airway organoid technology is needed, since greater variability in results was reported as compared to those of HBE and HNE cells in ALI cultures [26,47][43][44]. Airway organoids also exhibited CFTR-independent swelling that was promoted by the stimulation of alternative ion channels [49][46].2.3. Intestinal Organoids
Among PwCF, intestinal organoids have been frequently obtained from rectal biopsies, which is a relatively invasive procedure but one which is well tolerated by individuals [80][78]. From these samples, LGR5+ adult stem cells from intestinal crypts are isolated and cultured in a physical matrix—the most broadly used is matrigel—with a specific medium containing appropriate growth factors that enable their stemness maintenance for the expansion and self-organization into 3D structures termed organoids [81,82][79][80]. These cells can thus be cultured and expanded for long periods without losing their ability for self-renewal and growth. They can thus be used for the assessment of currently available modulators or be biobanked for future studies. Intestinal cells also have higher CFTR expression levels compared with airway cells [83][81], such as HBE and HNE cells, which offers an advantage for CFTR studies. Similar to airway organoids, the FIS is the most used assay for the assessment of CFTR modulators in intestinal organoids [80][78]. Since CFTR is active in healthy individuals, their organoids have a rounder shape, with a fluid-filled, steady-state lumen, under basal culture conditions. On the other hand, organoids from PwCF have a more irregular aspect with less visible lumen. Such differences led to the development of two scoring criteria for evaluating differences in organoid morphology: (i) the steady-state lumen area (SLA) [24,80][47][78] and (ii) the rectal organoid morphology analysis (ROMA) [84][82]. The SLA measures and compares the lumen area with the total organoid area, and is expressed as the percentage of the total organoid area [24,80][47][78]. The ROMA assesses the circularity index, which measures the roundness of the organoids, and the intensity ratio, which measures the presence/absence of a central lumen [84][82]. By using these parameters, both SLA and ROMA were able to discriminate between organoids of healthy individuals and PwCF, as well as CFTR rescue by modulator drugs [24,80,84][47][78][82]. Results from FIS of intestinal organoids demonstrated good correlations with responses in other samples from the same individual, namely current measurements in rectal biopsies and HNE cells [48,85][45][83]. Other studies have also demonstrated a strong correlation between FIS of intestinal organoids with SCC [24[47][48],50], which enabled the stratification of children with CF based on the disease severity [51][49]. Furthermore, ppFEV1 and body mass index presented consistent correlations with FIS of intestinal organoids [52,56][50][54]. Regarding CFTR modulators in PwCF, various studies demonstrated that the FIS assay of intestinal organoids can be a feasible biomarker for predicting clinical benefits [24,53,57][47][51][55]. Indeed, a high correlation between modulator-promoted responses in intestinal organoids and alterations in SCC and ppFEV1 in PwCF carrying common and rare variants has been reported in several studies [24,50,53][47][48][51]. Moreover, modulator-promoted responses in intestinal organoids have served as a basis for guiding eligibility for compassionate use and to obtain health insurance coverage for individuals carrying non-eligible responsive CFTR variants [54,58][52][56]. Altogether, these reports confirm the high throughput of organoids and support their use as a valuable tool for precision medicine approaches. However, limitations should be considered as these organoids are derived from intestinal cells and thus may not completely recapitulate airway/lung biology. Furthermore, although FIS of intestinal organoids appears to be very sensitive to CFTR function (even in a low functional range), assessments are limited to the structure stretching, which might underestimate high responses due to assay ceiling effects. Finally, organoids from healthy individuals are already pre-swollen in baseline conditions, indicating that FIS might also be underestimated in organoids of PwCF-carrying variants with high residual function.References
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662.
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462.
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065.
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073.
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080.
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723.
- Boon, M.; Verleden, S.E.; Bosch, B.; Lammertyn, E.J.; McDonough, J.E.; Mai, C.; Verschakelen, J.; Kemner-van de Corput, M.; Tiddens, H.A.W.; Proesmans, M.; et al. Morphometric Analysis of Explant Lungs in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 516–526.
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1.
- Sosnay, P.R.; White, T.B.; Farrell, P.M.; Ren, C.L.; Derichs, N.; Howenstine, M.S.; Nick, J.A.; De Boeck, K. Diagnosis of Cystic Fibrosis in Nonscreened Populations. J. Pediatr. 2017, 181S, S52–S57.e2.
- Cystic Fibrosis Mutation Database (CFTR1 Database). Available online: http://www.genet.sickkids.on.ca/ (accessed on 1 December 2023).
- Clinical and Function Translation of CFTR (CFTR2 Database). Available online: https://cftr2.org/ (accessed on 1 December 2023).
- Koch, C.; Cuppens, H.; Rainisio, M.; Madessani, U.; Harms, H.; Hodson, M.; Mastella, G.; Navarro, J.; Strandvik, B.; McKenzie, S.; et al. European Epidemiologic Registry of Cystic Fibrosis (ERCF): Comparison of major disease manifestations between patients with different classes of mutations. Pediatr. Pulmonol. 2001, 31, 1–12.
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38.
- Oliver, K.E.; Carlon, M.S.; Pedemonte, N.; Lopes-Pacheco, M. The revolution of personalized pharmacotherapies for cystic fibrosis: What does the future hold? Expert Opin. Pharmacother. 2023, 24, 1545–1565.
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34.
- Lopes-Pacheco, M.; Pedemonte, N.; Veit, G. Discovery of CFTR modulators for the treatment of cystic fibrosis. Expert Opin. Drug Discov. 2021, 16, 897–913.
- Ramsey, B.W.; Davies, J.C.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672.
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231.
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035.
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023.
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948.
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819.
- Bacalhau, M.; Camargo, M.; Magalhães-Ghiotto, G.A.V.; Drumond, S.; Castelletti, C.H.M.; Lopes-Pacheco, M. Elexacaftor-Tezacaftor-Ivacaftor: A Life-Changing Triple Combination of CFTR Modulator Drugs for Cystic Fibrosis. Pharmaceuticals 2023, 16, 410.
- Silva, I.A.L.; Laselva, O.; Lopes-Pacheco, M. Advances in Preclinical In Vitro Models for the Translation of Precision Medicine for Cystic Fibrosis. J. Pers. Med. 2022, 12, 1321.
- Caputo, A.; Hinzpeter, A.; Caci, E.; Pedemonte, N.; Arous, N.; Di Duca, M.; Zegarra-Moran, O.; Fanen, P.; Galietta, L.J.V. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J. Pharmacol. Exp. Ther. 2009, 330, 786–791.
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245.
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; De Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748.
- Fidler, M.C.; Buckley, A.; Sullivan, J.C.; Statia, M.; Boj, S.F.; Vries, R.G.J.; Munck, A.; Higgins, M.; Moretto Zita, M.; Negulescu, P.; et al. G970R-CFTR Mutation (c.2908G>C) Results Predominantly in a Splicing Defect. Clin. Transl. Sci. 2021, 14, 656–663.
- Pedemonte, N.; Tomati, V.; Sondo, E.; Galietta, L.J.V. Influence of cell background on pharmacological rescue of mutant CFTR. Am. J. Physiol. Cell Physiol. 2010, 298, C866–C874.
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885.
- Tomati, V.; Costa, S.; Capurro, V.; Pesce, E.; Pastorino, C.; Lena, M.; Sondo, E.; Di Duca, M.; Cresta, F.; Cristadoro, S.; et al. Rescue by elexacaftor-tezacaftor-ivacaftor of the G1244E cystic fibrosis mutation’s stability and gating defects are dependent on cell background. J. Cyst. Fibros. 2023, 22, 525–537.
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic fibrosis lung disease modifiers and their relevance in the new era of precision medicine. Genes 2021, 12, 562.
- McGarry, M.E.; McColley, S.A. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr. Pulmonol. 2021, 56, 1496–1503.
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 375.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Moncivaiz, J.D.; Ostmann, A.J.; Strecker, L.M.; Clancy, J.P. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight 2018, 3, e99385.
- Noel, S.; Servel, N.; Hatton, A.; Golec, A.; Rodrat, M.; Ng, D.R.S.; Li, H.; Pranke, I.; Hinzpeter, A.; Edelman, A.; et al. Correlating genotype with phenotype using CFTR-mediated whole-cell Cl− currents in human nasal epithelial cells. J. Physiol. 2022, 600, 1515–1531.
- Debley, J.S.; Barrow, K.A.; Rich, L.M.; Singh, P.; McKone, E.F.; Nichols, D.P. Correlation between ivacaftor-induced CFTR activation in airway epithelial cells and improved lung function: A proof-of-concept study. Ann. Am. Thorac. Soc. 2020, 17, 1024–1027.
- McGarry, M.E.; Illek, B.; Ly, N.P.; Zlock, L.; Olshansky, S.; Moreno, C.; Finkbeiner, W.E.; Nielson, D.W. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: N-of-1 studies. Pediatr. Pulmonol. 2017, 52, 472–479.
- Park, J.K.H.; Shrivastava, A.; Zhang, C.; Pollok, B.A.; Finkbeiner, W.E.; Gibb, E.R.; Ly, N.P.; Illek, B. Functional Profiling of CFTR-Directed Therapeutics Using Pediatric Patient-Derived Nasal Epithelial Cell Models. Front. Pediatr. 2020, 8, 536.
- Terlizzi, V.; Pesce, E.; Capurro, V.; Tomati, V.; Lena, M.; Pastorino, C.; Bocciardi, R.; Zara, F.; Centrone, C.; Taccetti, G.; et al. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators. Int. J. Mol. Sci. 2023, 24, 6576.
- Dreano, E.; Burgel, P.R.; Hatton, A.; Bouazza, N.; Chevalier, B.; Macey, J.; Leroy, S.; Durieu, I.; Weiss, L.; Grenet, D.; et al. Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 2023, 62, 2300110.
- Allan, K.M.; Astore, M.A.; Kardia, E.; Wong, S.L.; Fawcett, L.K.; Bell, J.L.; Visser, S.; Chen, P.-C.; Griffith, R.; Jaffe, A.; et al. Q1291H-CFTR molecular dynamics simulations and ex vivo theratyping in nasal epithelial models and clinical response to elexacaftor/tezacaftor/ivacaftor in a Q1291H/F508del patient. Front. Mol. Biosci. 2023, 10, 1148501.
- Guimbellot, J.S.; Leach, J.M.; Chaudhry, I.G.; Quinney, N.L.; Boyles, S.E.; Chua, M.; Aban, I.; Jaspers, I.; Gentzsch, M. Nasospheroids permit measurements of CFTR-dependent fluid transport. JCI Insight 2017, 2, e95734.
- Brewington, J.J.; Filbrandt, E.T.; LaRosa, F.J.; Ostmann, A.J.; Strecker, L.M.; Szczesniak, R.D.; Clancy, J.P. Detection of CFTR function and modulation in primary human nasal cell spheroids. J. Cyst. Fibros. 2018, 17, 26–33.
- Anderson, J.D.; Liu, Z.; Odom, L.V.; Kersh, L.; Guimbellot, J.S. CFTR function and clinical response to modulators parallel nasal epithelial organoid swelling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L119–L129.
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300.
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 2016, 8, 344ra84.
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.
- de Winter-De Groot, K.M.; Janssens, H.M.; van Uum, R.T.; Dekkers, J.F.; Berkers, G.; Vonk, A.; Kruisselbrink, E.; Oppelaar, H.; Vries, R.; Clevers, H.; et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur. Respir. J. 2018, 52, 1702529.
- de Winter-de Groot, K.M.; Berkers, G.; Marck-van der Wilt, R.E.P.; van der Meer, R.; Vonk, A.; Dekkers, J.F.; Geerdink, M.; Michel, S.; Kruisselbrink, E.; Vries, R.; et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J. Cyst. Fibros. 2020, 19, 614–619.
- Ramalho, A.S.; Förstová, E.; Vonk, A.M.; Ferrante, M.; Verfailli, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekma, J.M.; Sarouk, I.; et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 2021, 57, 1902426.
- Aalbers, B.L.; Brunsveld, J.E.; van der Ent, C.K.; van den Eijnden, J.C.; Beekman, J.M.; Heijerman, H.G.M. Forskolin induced swelling (FIS) assay in intestinal organoids to guide eligibility for compassionate use treatment in a CF patient with a rare genotype. J. Cyst. Fibros. 2022, 21, 254–257.
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of organoid swelling and in vivo biomarkers of cftr function to determine effects of lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for the f508del mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592.
- Muilwijk, D.; de Poel, E.; van Mourik, P.; Suen, S.W.F.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; Oppelaar, H.; Hagemeijer, M.C.; Berkers, G.; et al. Forskolin-induced Organoid Swelling is Associated with Long-term CF Disease Progression. Eur. Respir. J. 2022, 60, 2100508.
- Lefferts, J.W.; Bierlaagh, M.C.; Kroes, S.; Nieuwenhuijze, N.D.A.; Sonneveld van Kooten, H.N.; Niemöller, P.J.; Verburg, T.F.; Janssens, H.M.; Muilwijk, D.; van Beuningen, S.F.B.; et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. Int. J. Mol. Sci. 2023, 24, 14539.
- Mitropoulou, G.; Brandenberg, N.; Hoehnel, S.; Ceroni, C.; Balmpouzis, Z.; Blanchon, S.; Dorta, G.; Sauty, A.; Koutsokera, A. Rectal organoid-guided CFTR modulator therapy restores lung function in a CF patient with the rare 1677delTA/R334W genotype. Eur. Respir. J. 2022, 60, 2201341.
- Kondratyeva, E.; Melyanovskaya, Y.; Efremova, A.; Krasnova, M.; Mokrousova, D.; Bulatenko, N.; Petrova, N.; Polyakov, A.; Adyan, T.; Kovalskaia, V.; et al. Clinical and Genetic Characteristics of a Patient with Cystic Fibrosis with a Complex Allele and Functional Evaluation of the CFTR Channel. Genes 2023, 14, 1705.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830.
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848.
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620.
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121.
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607.
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231.
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451.
- Müller, L.; Brighton, L.E.; Carson, J.L.; Fischer, W.A.; Jaspers, I. Culturing of human nasal epithelial cells at the air liquid interface. J. Vis. Exp. 2013, 80, e50646.
- Sette, G.; Lo Cicero, S.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping cystic fibrosis in vitro in ALI culture and organoid models generated from patient-derived nasal epithelial conditionally reprogrammed stem cells. Eur. Respir. J. 2021, 58, 2100908.
- Wu, X.; Wang, S.; Li, M.; Li, J.; Shen, J.; Zhao, Y.; Pang, J.; Wen, Q.; Chen, M.; Wei, B.; et al. Conditional reprogramming: Next generation cell culture. Acta Pharm. Sin. B 2020, 10, 1360–1381.
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324.
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381.
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289.
- McDougall, C.M.; Blaylock, M.G.; Douglas, J.G.; Brooker, R.J.; Helms, P.J.; Walsh, G.M. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am. J. Respir. Cell Mol. Biol. 2008, 39, 560–568.
- Li, H.; Sheppard, D.N.; Hug, M.J. Transepithelial electrical measurements with the Ussing chamber. J. Cyst. Fibros. 2004, 3 (Suppl. S2), 123–126.
- Sheppard, D.N.; Gray, M.A.; Gong, X.; Sohma, Y.; Kogan, I.; Benos, D.J.; Scott-Ward, T.S.; Chen, J.-H.; Li, H.; Cai, Z.; et al. The patch-clamp and planar lipid bilayer techniques: Powerful and versatile tools to investigate the CFTR Cl− channel. J. Cyst. Fibros. 2004, 3, 101–108.
- Graeber, S.Y.; Balázs, A.; Ziegahn, N.; Rubil, T.; Vitzthum, C.; Piehler, L.; Drescher, M.; Seidel, K.; Rohrbach, A.; Röhmel, J.; et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 12365.
- Pranke, I.; Hatton, A.; Masson, A.; Flament, T.; Le Bourgeois, M.; Chedevergne, F.; Bailly, C.; Urbach, V.; Hinzpeter, A.; Edelman, A.; et al. Might brushed nasal cells be a surrogate for CFTR modulator clinical response? Am. J. Respir. Crit. Care Med. 2019, 199, 123–126.
- Calucho, M.; Gartner, S.; Barranco, P.; Fernández-Álvarez, P.; Pérez, R.G.; Tizzano, E.F. Validation of nasospheroids to assay CFTR functionality and modulator responses in cystic fibrosis. Sci. Rep. 2021, 11, 15511.
- Liu, Z.; Anderson, J.D.; Deng, L.; Mackay, S.; Bailey, J.; Kersh, L.; Rowe, S.M.; Guimbellot, J.S. Human nasal epithelial organoids for therapeutic development in cystic fibrosis. Genes 2020, 11, 603.
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945.
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265.
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772.
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289.
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal organoid morphology analysis (ROMA) as a promising diagnostic tool in cystic fibrosis. Thorax 2021, 76, 1146–1149.
- Silva, I.A.L.; Railean, V.; Duarte, A.; Amaral, M.D. Personalized Medicine Based on Nasal Epithelial Cells: Comparative Studies with Rectal Biopsies and Intestinal Organoids. J. Pers. Med. 2021, 11, 421.
More