Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Miquéias Lopes-Pacheco and Version 2 by Sirius Huang.
The implementation of cystic fibrosis (CF) transmembrane conductance regulator (CFTR) modulator drugs into clinical practice has been attaining remarkable therapeutic outcomes for CF, a life-threatening autosomal recessive genetic disease. However, there is elevated CFTR allelic heterogeneity, and various individuals carrying (ultra)rare CF genotypes remain without any approved modulator therapy. Novel translational model systems based on individuals’ own cells/tissue are now available and can be used to interrogate in vitro CFTR modulator responses and establish correlations of these assessments with clinical features, aiming to provide prediction of therapeutic effectiveness.
- airway cells
- bioassay
- biomarkers
- extracellular vesicles
- inflammation
- microRNA
- precision medicine
- organoids
- theratyping
1. Introduction
Inherited in an autosomal recessive pattern, cystic fibrosis (CF) is a life-threatening progressive disease affecting over 100,000 people worldwide [1][2][1,2]. The disease occurs due to mutations in the gene encoding the CF transmembrane conductance (CFTR) protein [3][4][5][3,4,5], a chloride/bicarbonate channel expressed at the apical plasma membrane (PM) that plays a vital role in regulating fluid and ion movements across several epithelial tissues, including lungs, intestine, pancreas, and sweat glands [1][6][1,6]. However, despite the multiorgan features of the disease, the elevated morbidity and mortality of people with CF (PwCF) are linked to an accelerated decline of lung function due to repeated cycles of airway mucus accumulation, chronic inflammation, and persistent infection that lead to tissue remodeling and respiratory failure [6][7][6,7].
CF diagnosis is based on symptomatology consistent with the disease and laboratory biomarkers that provide evidence of CFTR dysfunction [8][9][8,9]. Most new cases of CF are nowadays identified within newborn screening programs by assessing immunoreactive trypsinogen in bloodspots. To confirm the diagnosis, CFTR (dys)function should be demonstrated by: (i) identification of CFTR pathogenic variants in both alleles by genetic analysis, and (ii) elevated sweat chloride concentration (SCC; >60 mmol·L−1), altered transepithelial nasal potential difference (NPD) and/or intestinal current measurement (ICM) [8][9][8,9]. Although SCC is the standard test for CF diagnosis, the alternative biomarkers are particularly relevant in cases in which clinical signs and symptoms are suggestive of CF, but SCC is intermediate (30–59 mmol·L−1) [8][9][8,9].
Over 2100 variants have been identified in the CFTR gene [10], of which approximately one-third are now classified as CF-causing [11]. The deletion of a phenylalanine at position 508 (p.Phe508del, legacy: F508del) is the most prevalent CF-causing variant, accounting for approximately 70% of all CF alleles [1], while the remaining 30% of CF alleles are represented by an enormous number of CFTR variants and most are (ultra)rare, occurring among few PwCF worldwide [11]. Due to such CFTR allelic heterogeneity, distinct CF phenotypes exist—on average, PwCF with pancreatic insufficient exhibit more severe forms of the disease, while milder phenotypes are usually associated with pancreatic sufficiency [12]. Indeed, these variants cause distinct primary defects, comprising CFTR mRNA and protein biosynthesis, anion transport, and/or PM turnover. Therefore, they have been separated into CFTR variant classes, which are characterized by alterations in (I) expression, (II) folding and trafficking, (III) gating, (IV) conductance, (V) abundance, and (VI) PM stability [13][14][13,14]. Overall, CFTR variants in classes I and II are associated with a minimal (or null) function, while a residual (or some) function is usually observed in those variants in classes IV–VI. This grouping offers the advantage that CFTR variants with similar defects might be tackled by similar therapeutic strategies—i.e., theratyping [15].
Over the last two decades, precision (or personalized) medicines targeting the fundamental cause of CF have been developed with tremendous accomplishments attained [14][16][14,16]. Indeed, four CFTR modulator drugs are now approved for clinical use: the correctors lumacaftor (LUMA, VX-809), tezacaftor (TEZA, VX-661), and elexacaftor (ELX, VX-445), which retrieve CFTR folding and trafficking to the PM, and the potentiator ivacaftor (IVA, VX-770), which enhances CFTR channel open probability. These drugs—particularly the ‘highly effective’ ones—have provided impressive clinical benefits, representing thus a new dawn for PwCF with eligible genotypes [17][18][19][20][21][22][23][17,18,19,20,21,22,23]. However, there is a significant number of PwCF carrying (ultra)rare variants for whom no modulator therapy has been approved [11][16][11,16].
 While cells heterologously expressing CF-causing variants remain very useful for CFTR studies, primary cell models can provide a more sensitive and reliable prediction of therapeutic responses for several reasons: (i) The last express CFTR in the native genomic context, while cell lines frequently use CFTR cDNA (a copy of the mature mRNA, which lacks the introns); therefore, cells lines may not recapitulate certain cellular mechanisms, including nonsense-mediated decay or splicing effects. For instance, p.Gly970Arg (legacy G970R) was thought to be a CFTR gating variant based on cDNA expression findings [25][26][30,31]; however, analysis of cells from PwCF carrying this variant revealed that it actually causes a splicing defect [27][28][32,33]. (ii) The cellular background has a marked influence on CFTR processing and function as well as its pharmacological sensitivity. As exemplified by the cases of p.Phe508del [29][30][34,35] and p.Gly1244Glu [31][36], the complexity of cellular processes related to protein biogenesis and folding, as well as its PM trafficking, may not be completely recapitulated in cell lines—particularly those from non-human and/or non-respiratory epithelium origin [24][29]. Likewise, CFTR gene regulation can be impacted by epigenetic factors and these are only taken into account by the assessment in primary cells from each individual [32][37]. (iii) Characterization of single variants can be efficiently accomplished in cell lines; however, two variants indicating to be low responsive (below therapeutic relevant threshold) in separated cell lines can compose a genotype with a good prediction for clinical benefits (if evidenced by assessing responses in this individual’s cells). This is particularly relevant for (ultra)rare CFTR variants that are frequently identified in racial and ethnic minority populations, which are usually excluded from traditional clinical trial designs [33][38].
Moreover, numerous reports have established correlations of the data on primary cell models with clinical features of PwCF (before and after initiating modulator therapy) to provide a translational perspective of therapeutic effects (Table 1). Accordingly, these can serve as potential biomarkers to identify which drug(s) could be the best therapeutics for every individual with CF—i.e., “the right therapy for the right person”.
While cells heterologously expressing CF-causing variants remain very useful for CFTR studies, primary cell models can provide a more sensitive and reliable prediction of therapeutic responses for several reasons: (i) The last express CFTR in the native genomic context, while cell lines frequently use CFTR cDNA (a copy of the mature mRNA, which lacks the introns); therefore, cells lines may not recapitulate certain cellular mechanisms, including nonsense-mediated decay or splicing effects. For instance, p.Gly970Arg (legacy G970R) was thought to be a CFTR gating variant based on cDNA expression findings [25][26][30,31]; however, analysis of cells from PwCF carrying this variant revealed that it actually causes a splicing defect [27][28][32,33]. (ii) The cellular background has a marked influence on CFTR processing and function as well as its pharmacological sensitivity. As exemplified by the cases of p.Phe508del [29][30][34,35] and p.Gly1244Glu [31][36], the complexity of cellular processes related to protein biogenesis and folding, as well as its PM trafficking, may not be completely recapitulated in cell lines—particularly those from non-human and/or non-respiratory epithelium origin [24][29]. Likewise, CFTR gene regulation can be impacted by epigenetic factors and these are only taken into account by the assessment in primary cells from each individual [32][37]. (iii) Characterization of single variants can be efficiently accomplished in cell lines; however, two variants indicating to be low responsive (below therapeutic relevant threshold) in separated cell lines can compose a genotype with a good prediction for clinical benefits (if evidenced by assessing responses in this individual’s cells). This is particularly relevant for (ultra)rare CFTR variants that are frequently identified in racial and ethnic minority populations, which are usually excluded from traditional clinical trial designs [33][38].
Moreover, numerous reports have established correlations of the data on primary cell models with clinical features of PwCF (before and after initiating modulator therapy) to provide a translational perspective of therapeutic effects (Table 1). Accordingly, these can serve as potential biomarkers to identify which drug(s) could be the best therapeutics for every individual with CF—i.e., “the right therapy for the right person”.
2. Laboratory Tools to Predict CFTR Modulator Effectiveness In Vivo
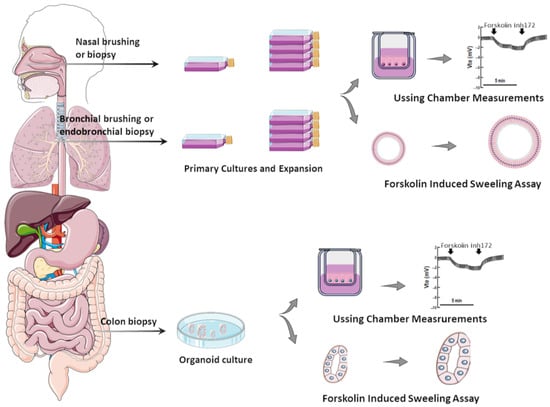
Several in vitro assays have been developed using various model systems to comparatively assess the efficacy of CFTR modulators (individually or combinations thereof) [24][29]. Although heterologous cell lines have been fundamental to enhance the understanding of CFTR biology at genetic, biochemical, and physiological levels, in the context of precision medicine, they can only be used for variant theratyping (i.e., matching single variants to modulators), being unable to predict responses of a determined individual to a specific therapy. Accordingly, alternative translational models have been established by using primary cells from PwCF to inquire about responses at an individual level (Figure 1).Figure 1. Translational human CF model systems for personalized/precision medicine. Airway samples can be collected from the nose or bronchi by brushing or biopsies and gastrointestinal samples can be collected from the colon or rectum by biopsies. These samples are cultured in specific in vitro conditions, expanded, and then seeded on porous membranes to grow in polarized monolayers or in matrigel to form organoids/spheroids. Cell monolayers and organoids can be used to assess CFTR function/rescue through Ussing chamber measurements and through forskolin-induced swelling assays, respectively.
Table 1.
Studies reporting correlations of CFTR modulator-promoted responses in cell models and clinical effects in PwCF.
| Model System | CF Population/Genotypes | CFTR Modulator(s) | Key Findings/Correlations | Ref. |
|---|---|---|---|---|
| HNE cells | PwCF homozygous for p.Phe508del or carrying genotypes leading to minimal or residual CFTR function | LUMA TEZA IVA |
The level of CFTR rescue significantly correlated with the ppFEV1 change at 6 months in 8 PwCF treated with CFTR modulators. | [34][25] |
| Ten PwCF (<19 years) with a wide range of CFTR variants | LUMA IVA |
CFTR modulation produced changes in CFTR function in nasal and bronchial cell cultures. There was a correlation between the residual CFTR function in both cell types from PwCF with SCC between 60 and 90 mmol·L−1. |
[35][39] | |
| Five healthy subjects (they were not genetically tested) and eight PwCF with variants associated with minimal or residual CFTR function | – | CFTR-mediated transepithelial chloride currents measured in vitro through Ussing chamber assay correlated with the SCC of subjects. | [36][40] | |
| Twelve adults with CF with either the p.Gly551Asp or p.ArgR117His | IVA | A strong correlation between changes in SCC and in vitro CFTR activation was observed. Furthermore, a moderate correlation between in vitro CFTR activation and changes in ppFEV1 was reported. | [37][41] | |
| Seven PwCF (≤16 years, clinically stable) with residual CFTR function variants |
IVA | All subjects with decreased SCC in response to IVA treatment also had significant increases in chloride current in HNE cultures with IVA exposure. | [38][42] | |
| Healthy volunteers, PwCF with p.PheF508del/p.Arg117His-7T, p.PheF508del/p.PheF508del, p.PheF508del/p.Met284fs, p.Arg334Trp/c.406-1G>A, and p.Ser18ArgfsX16/p.Ser18ArgfsX16 | LUMA TEZA IVA |
In vitro CFTR chloride measurements correlated to changes in SCC. | [39][43] | |
| Five PwCF: one homozygous for p.Ser737Phe and four compound heterozygous for p.Ser737Phe | TEZA ELX IVA |
In vitro analysis demonstrated different levels of CFTR activity. Some degree of CFTR dysfunction was detected by evaluating chloride secretion in HNE cells derived from compound heterozygous subjects; CFTR activity was improved by IVA alone and even more by treatment with correctors. | [40][44] | |
| Eleven PwCF carrying FDA-eligible variants for ELX/TEZA/IVA and twenty-eight PwCF carrying non-eligible variants | TEZA ELX IVA |
There was a significant relationship between CFTR activity correction and changes in ppFEV1 or SCC. | [41][45] | |
| A 56-year-old male with CF and the p.Phe508del/p.Gln1291His genotype | TEZA ELX IVA |
In HNE cells, p.Phe508del/p.Gln1291His resulted in reduced baseline CFTR activity, and showed minimal response to ELX/TEZA/IVA (individually or in combination), aligning with the individual’s clinical evaluation as a non-responder to these drugs. | [42][46] | |
| Airway organoids | Nine healthy volunteers and three PwCF | LUMA IVA |
Organoids treated with modulators displayed similar effects to clinical response; LUMA/IVA treatment promoted greater responses in organoids from p.PheF508del-homozygous subjects. | [43][26] |
| Six healthy volunteers, nine PwCF homozygous for p.Phe508del, and ten with at least one non-p.Phe508del variant | LUMA IVA |
p.Phe508del/p.Phe508del organoids treated with LUMA/IVA exhibited a positive change in swelling responses; a relationship between organoid response to drug and in vivo clinical response was observed for three subjects treated. | [44][47] | |
| Eighteen PwCF and five healthy volunteers | TEZA ELX IVA |
In vitro responses to CFTR modulators correlated well with clinical measurements. | [45][48] | |
| Five PwCF: p.Phe508del/p.Phe508del, p.Phe508del/p.Gly542X, and F508del/G542X and p.Arg334Trp/p.Arg334Trp | LUMA IVA |
Responses of organoids correlated well with clinical findings. | [46][49] | |
| Intestinal organoids | Twelve healthy volunteers, four CF carriers (WT/p.Phe508del), thirty-five PwCF carrying class II and III variants, and eighteen PwCF carrying class IV and V variants |
LUMA IVA |
Responses of organoids to CFTR modulators correlated with outcome data from clinical trials. | [47][24] |
| Twenty-four PwCF: fifteen carrying at least one p.Ser1251Asn and nine carrying at least one (ultra)rare CFTR variant | LUMA IVA |
Responses to CFTR modulators in organoids correlated with in vivo measurements (SCC and ppFEV1). | [48][50] | |
| Thirty-four children with CF carrying a wide range of CFTR variants | – | Organoid swelling significantly correlated with SCC and ICM. | [49][51] | |
| Thirty-four PwCF with p.Phe508del/p.Phe508del | – | Variability in CFTR residual function appeared to contribute to the clinical heterogeneity and organoid swelling values; responses in organoids correlated with ppFEV1 and BMI. | [50][52] | |
| Ninety-seven PwCF with well-characterized and rare CFTR variants | LUMA IVA |
Measurements of residual CFTR function and rescue by CFTR modulators in organoids correlated with clinical data, namely changes in ppFEV1 and SCC. | [51][53] | |
| A 56-year-old female with CF carrying p.Phe508del/c.3717+5G>T | LUMA TEZA IVA |
No baseline swelling was observed in organoids, suggesting minimal CFTR function; however, limited swelling was detected after LUMA/IVA or TEZA/IVA treatment. | [52][54] | |
| Twenty-one PwCF homozygous for p.Phe508del | LUMA IVA |
No correlations were found between organoid swelling and changes in the in vivo biomarkers, namely SCC and NPD. | [53][55] | |
| A total of 173 PwCF carrying a wide range of CFTR variants | – | Organoid swelling values were associated with long-term ppFEV1 decline and the probability of developing different CF-related comorbidities, namely pancreatic insufficiency, CF-related liver disease, and CF-related diabetes. | [54][56] | |
| Fifteen PwCF homozygous for p.Phe508del, fifteen PwCF carrying p.Phe508del/class I variant, and twenty-two PwCF with rare variant non-eligible for CFTR modulator therapy | TEZA ELX IVA |
Responses to modulators in organoids from p.Phe508del/p.Phe508del or p.Phe508del/class I variant correlated with changes in ppFEV1. In CF organoids with 11 rare genotypes, CFTR function restoration was reported upon ELX/TEZA/IVA treatment. |
[55][57] | |
| A 19-year-old female with CF carrying p.Tyr515X/p.Arg334Trp | LUMA TEZA ELX IVA |
Organoids treated with IVA alone or in combination with correctors demonstrated similar rescue of CFTR-dependent fluid secretion. In vivo measurements demonstrated significant clinical improvements in SCC, ppFEV1, and respiratory symptoms after 7 days of IVA initiation that were sustained for the 9-month follow-up. |
[56][58] | |
| A 6-year-old male with CF carrying p.Phe508del/p.Glu217Gly-Gly509Asp | LUMA TEZA ELX IVA |
The p.Glu217Gly-Gly509Asp complex allele was characterized by a high residual function of the CFTR channel; organoid swelling values demonstrated rescue of CFTR function by all tested modulators. | [57][59] |
Abbreviations: BMI—body mass index; CF—cystic fibrosis; CFTR—CF transmembrane conductance regulator; ELX—elexacaftor; HNE—human nasal epithelial; ICM—intestinal current measurement; IVA—ivacaftor; LUMA—lumacaftor; NPD—nasal potential differences; PwCF—people with CF; ppFEV1—percent predicted forced expiratory volume in 1 s; SCC—sweat chloride concentration; TEZA—tezacaftor.