Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Miquéias Lopes-Pacheco and Version 2 by Peter Tang.
Sepsis and acute respiratory distress syndrome (ARDS) are complex and heterogeneous syndromes for which no specific therapies exist. Mesenchymal stromal cell (MSC) administration significantly reduces tissue inflammation and remodeling, improves pathogen clearance, and reduces morbidity and mortality in multiple preclinical models of sepsis and acute lung injury (ALI, the animal corollary to ARDS)
- acute lung injury
- acute respiratory distress syndrome
- extracellular vesicles
- exosomes
- mesenchymal stromal cells
- microRNA
- nano therapy
- sepsis
1. Introduction
Sepsis and acute respiratory distress syndrome (ARDS) are complex and heterogeneous syndromes for which no specific therapies exist. Mesenchymal stromal cell (MSC) administration significantly reduces tissue inflammation and remodeling, improves pathogen clearance, and reduces morbidity and mortality in multiple preclinical models of sepsis and acute lung injury (ALI, the animal corollary to ARDS) [1]. Significant morbidity and mortality associated with both syndromes, as well as the lack of specific therapies, explains the urgency in demonstrating the therapeutic benefit of allogenic delivery of MSCs in the clinic. Although multiple studies have identified various targets that, when pharmacologically or interventionally altered, mitigate various pathological features of sepsis and ARDS experimentally (in vitro and in vivo) and in Phase I and II clinical trials, none has yielded effective treatments in Phase III randomized clinical trials.
Since the initial isolation and culture of human MSCs over 1300 registered clinical trials (clinicaltrials.gov “mesenchymal” 6/5/20) [2] have demonstrated the safety of administering these cells in humans [3]. Notwithstanding, we have yet to replicate the large effect sizes predicted from pre-clinical research [4][5][4,5], as small and large trials have failed to meet efficacy endpoints [6]. Inconsistent results have been attributed to product heterogeneity and irregularities, use of pre-clinical models that may not recapitulate the complexities of human disease, transferability across species, and/or poor estimation of effect size from preclinical data leading to inconclusive findings in humans [2][7][2,7]. Moreover, although the potential of MSCs remains undisputed, questions remain concerning their mechanisms of action (MoA).
2. MSCs Are Not Required for Therapeutic Effect in Sepsis/ALI
In preclinical studies of sepsis and ALI, MSCs delivered through a variety of different routes moderate multiple organ failure and reduce mortality [1]. From the initial studies, various investigators argued that although the precise MoA remains to be elucidated, cell engraftment with differentiation, transdifferentiation, or cell fusion were unlikely to contribute to observed beneficial effects in preclinical models of sepsis/ALI. First, very low levels of cell persistence could be demonstrated after infusion. Following intravenous administration, MSCs are efficiently targeted to the lung where they are trapped simply by size-based filtration in the distal pulmonary arteriolar bed [8][10]. Using highly sensitive methods (i.e., quantitative real-time polymerase chain reaction (RT-PCR) for the sex-determining region Y protein in female recipients [8][9][10][10,11,12]) or fluorescent labeling approaches [11][12][13][13,14,15], cells seem to be present in the lung only up to 28–96 h post-infusion. Second, whole cells are not required for their therapeutic effect. Over a decade ago, it became apparent that the cultured supernatant from MSCs can mitigate lung injury by providing similar protection to that observed with viable whole cells [14][15][22,23], even when aerosolized [16][24]. In addition to confirming cell homing is not necessary for imparting therapeutic effects [17][25], this finding strongly supported a paracrine MoA. Multiple studies have also convincingly demonstrated that the immunomodulatory effect of MSCs is communicated via MSC and recipient-secreted cytokines and relies on the local microenvironment [18][19][20][21][22][19,26,27,28,29], as some of the observed effects depend on a pre-treatment of MSCs with inflammatory cytokines [20][21][23][27,28,30]. More recent findings indicate that the cytokine-mediated effects are only one part of the equation, as MSC-derived microparticles (including exosomes), apoptotic, metabolically inactivated, or even fragmented MSCs and MSC-membranes have immunomodulatory potential [24][25][26][27][28][31,32,33,34,35]. These observations have raised important questions regarding the putative direct targets of MSCs and the active therapeutic “ingredients” contained in an MSC, its secretome-derived “soup” or/and -derived microparticle/fragments, that confer their beneficial effects.3. MSC-Derived Secretome
A large portion of MSC’s therapeutic activity is attributed to direct primary signaling through their secretome, comprising a multitude of cytokines, chemokines, growth factors, and subcellular vesicles; overviews of the current state of knowledge of MSC’s secreted mediators and how inflammatory priming influences their release have been published [29][30][31][36,37,38]. Target identification studies have demonstrated that MSCs have a wide pleiotropic effect on the pathophysiology of complex syndromes, specifically sepsis and ALI. The Ouresearchers' group has previously published—using a model of polymicrobial sepsis induced by cecal ligation and puncture (CLP)—that MSC administration alters the expression of 3968 genes in five different sepsis-target organs (lung, liver, spleen, kidneys, and heart) [32][39]. Given about 30,000 as the estimated total number of genes in the mouse genome, theour data suggest that approximately 13% of the septic genome is transcriptionally reprogrammed after MSC administration [32][39]. While multiple gain- (overexpressing) and loss- (silencing)-of-function studies have identified key mediators involved in the therapeutic effect, including IL-10 [33][40] and anti-bacterial peptide LL-37 [34][41], none have emerged as the single most responsible therapeutic target. The release of EVs in the secretome represents an immediate cell communication mechanism that fulfills major criteria to impart MSC-related functions: (a) dissemination of membrane-bound mediators/signaling molecules; (b) transfer of expression patterns from one cell to another; and (c) rapid rearrangement of the cell surface. Like all messengers, EVs can not only immediately modify a phenotype of neighboring cells in a paracrine fashion but also bring forward an altered micro-milieu to other tissues by transferring the membrane composition and/or expression pattern to distal organs [35][42]. Salutary as well as pathological effects may be imparted by information contained on EVs and their cargo in the form of circulating nucleic acids (mRNAs and miRs), lipids, and proteins [24][36][37][31,43,44]. Seminal work from Monsel and colleagues demonstrated the therapeutic effects of human MSC-derived EVs in a mouse model of severe pneumonia [38][45]. In this model, the group was able to show that EV delivery significantly enhanced the expression of keratinocyte growth factor (KGF) in recipients associated with improved survival. A subsequent clinical trial randomized patients with ARDS to receive recombinant KGF or placebo. The trial was stopped early because of increased harm in the group that received KGF [39][46]. While multiple reasons are usually linked to unsuccessful clinical trials, results did suggest that the increase in KGF is a meta-phenomenon rather than directly causal to the improved mortality seen in animal models. Herein lies the difficulty and the critical importance of understanding and translating promising preclinical data to the clinic. Together, these data challenge the paradigm that a single, specific MSC-derived paracrine mediator is responsible for the global pleiotropic effect of MSCs on transcriptional and network reprogramming in sepsis. A much more plausible explanation is that MSC-conferred protection from sepsis includes a range of complementary activities, resulting in the mitigation of the innate and acquired immune and inflammatory responses. Of particular interest to the ouresearchers' group is the potential therapeutic role of regulatory RNAs contained within EVs. Below, the researchers rewe reviewed the literature implicating epigenetic regulatory small RNAs, specifically miRs, as important regulators and mediators of the therapeutic effect of MSC-derived EVs.4. MSC-EV Content
MSC-derived EVs act as paracrine mediators of MSCs’ beneficial effects. In a landmark publication [40][47], Phinney and colleagues demonstrated that MSCs are inefficient in performing mitophagy; using microvesicles to shuttle mitochondrial components out of the cell. As MSCs outsource mitophagy to recipient macrophages [40][47], they simultaneously package anti-inflammatory miRs into smaller EVs (including microRNA let-7f, c and I, 23b, 27b, and 29a). These miRs were shown by the group to inhibit critical pattern and damage recognition receptors and signaling molecules: Toll-like receptor (TLR)4, MyD88, TLR9, TLR7, and tumor necrosis factor-alpha (TNF-α) in recipient macrophages. There is little doubt EVs contain anti-inflammatory miRs, that are produced and released by MSCs; what is unclear is why the cells would do this, and whether these miRs are actually therapeutically active? Much of the controversy around which component of EV payload is biologically active (contributes to MoA) comes from non-standardized EV preparations. The importance of this issue cannot be understated [41][42][43][58,59,60]. The International Society for EVs has established clear criteria for isolating, characterizing, and defining microparticles derived from MSCs [44][45][61,62]. Moreover, Dr. Sai Kiang Lim and colleagues have recently published criteria for defining MSC-derived EVs for therapeutic application, including establishing MSC origin (concentration of CD73, CD90, and CD105; and absence of non-MSC antigens CD14, CD34, and CD11b); number of particles per unit weight protein/membrane lipids and particle diameter (within 50–200 nm); molar/weight ratio of protein to membrane lipids (e.g., cholesterol and phosphatidylcholine); and presence of biochemically active cargo (enzyme activity of CD73, unit activity per μg protein) [46][63]. The fact that MSC-derived EVs and miRs are therapeutically relevant is not disputed; the issue is whether the miRNAs that are carried inside EVs are actually the ones imparting their beneficial effects.5. EV-microRNAome
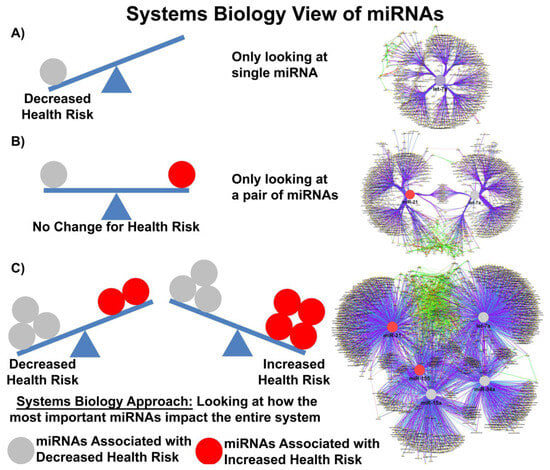
In the publication from Valadi and others, the authors determined that each exosome may carry as many as 100–120 different miRs and that some miRs were expressed to a greater extent in exosomes compared to donor cells [47][48]. Baglio and colleagues further demonstrated that MSCs of different origins have similar small RNA expression profiles; EV-derived miRs (EV-miRs) or exo-miRs represent about 1–2% of the cellular RNA content, and multiple highly expressed miRs are precluded from EV sorting [48][64], suggesting that the process of packaging miRs into EVs is highly regulated and not simply a mechanism for managing cellular waste. Quantitative and stoichiometric analysis of the miR content of EVs demonstrated less than one copy of each microRNA per EV [49][65]. Using ultrasensitive total internal reflection-based single-vesicle in situ quantitative and stoichiometric analysis of tumor-derived EV miRs, He and colleagues demonstrated that each EV may contain as many as 6 copies of different unique microRNAs [50][66]. But what is the functional dose of a miR? Based on the literature, the functional dose may differ for each target [51][67]; with factors such as the expression levels of the targets, the number of miR-binding sites on the target mRNA and feedback loops able to shift this narrow window for effective gene suppression. MiRs may also repress their targets in a nonlinear manner introducing thresholds in gene expression [52][68]. Furthermore, as miRs can work cooperatively or competitively [53][69], dose-dependent mRNA target selection becomes even more complicated when combinations of miRs, with potentially overlapping targets, are taken into account, such as would happen in EVs carrying a variety of different miRs. Electroporation nano straw delivery, which can bypass biological mechanisms, has been used to deliver miRs with precise dosage control directly into primary cells [54][70]. An example of the importance of dose comes from studies using miR-17-92, which was shown to decrease the cell viability of colon cancer cell line at low doses (0.00003 μg plasmid) but increase cell viability at high doses (0.3 μg plasmid) [51][67]. MiR fold changes as low as 3–4 are sufficient to drive the development of disease in transgenic mice [55][71], and a 1.5-fold change in the expression level of a single miR is considered enough to alter phenotype. Denzler and colleagues showed that when a miR is lowly expressed, only the highest-affinity sites are sufficiently occupied to mediate repression, but as miR expression increases, more and more intermediate and low-affinity sites have occupancies sufficient to mediate repression [56][72]. Canonical 6-nucleotide (-nt) sites, which typically mediate modest repression, can nonetheless compete for miR binding, with a potency of 20% of that observed for canonical 8-nt sites. This suggests there is a strong relationship between site affinity and miR-dose that is significantly impacted by competition between different miRs for overlapping binding sites. Moreover, cooperative binding of proximal sites for the same or different miRs does increase potency, making it very difficult to understand the relationship between dose and biological effect. In most clinical studies, EV doses range between 1–10 × 108–12 particles/mL, suggesting that while the copy number inside an EV may be small, the EV dose may deliver a significant number of therapeutically relevant miRs. Moreover, given that a single miR can regulate the expression of hundreds of genes, a system biologist’s view of miR networks suggests that the biological effects of a single functioning miR may significantly amplify the biological effect at a systems level (Figure 1) [57][73].Figure 1. Systems biology view of microRNA Networks. Figure generated with Cytoscape and ClueGO by Dr. Afshin Beheshti, Bioinformatician and Principal Investigator, Blue Marble Space Institute of Science, NASA Ames Research Center, Space Biosciences Research Branch, reproduced with permission from Dr. Beheshti and NASA Ames Research Center.