Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Niraj Neupane and Version 2 by Jason Zhu.
Breast cancer is a common type of cancer among women. One type, estrogen receptor-positive (ER+), is treated with endocrine therapies. However, some patients develop resistance to these therapies, which is a challenge. Scientists have developed second-generation drugs called selective estrogen receptor degraders (SERDs) that can overcome the limitations of the existing treatment.
- breast cancer
- estrogen receptor-positive
- endocrine therapies
- resistance
1. Mechanism of Action of SERDs
The activity of the ER is primarily mediated through its interaction with specific DNA sequences known as estrogen response elements (EREs). The estrogen binding to the ER promotes the dimerization of the receptor and its subsequent binding to the EREs located in the promoter regions of estrogen-responsive genes. Once bound to the ERE, the ER acts as a transcription factor, recruiting coactivators or corepressors to initiate the transcription of the target genes [1][9]. AIs primarily act by reducing the estrogen levels available for binding to the ER, thereby decreasing its activity as a transcription factor. In contrast, SERMs bind to the ER and modulate its activity in a tissue-specific manner. For example, in breast tissue, SERMs such as tamoxifen act as antagonists, competing with estrogen for the ER binding site and preventing the activation of genes involved in cell growth and proliferation. Selective estrogen receptor degraders (SERDs) are a newer class of ET that bind to the ER and induce its degradation, thereby reducing the overall activity of the receptor. Unlike SERMs, which can act as either agonists or antagonists, SERDs act exclusively as antagonists of the ER. This class of drugs includes fulvestrant, the first and, until January 2023, the only FDA-approved SERD. SERDs can block the effects of estrogen on breast cancer cells by binding to the ER and promoting its degradation, leading to a decrease in the expression and activity of ER target genes [2][11] (Figure 1). Fulvestrant has been shown to induce the proteasomal degradation of the ER in a dose-dependent manner [3][12]. Proteasomal degradation of an ER is achieved through its ability to promote the dissociation of chaperone proteins such as heat shock protein 90 (Hsp90) and p23 from the ER, resulting in the exposure of a hydrophobic surface on the receptor recognized by the E3 ubiquitin ligase complex. The E3 ligase then adds ubiquitin molecules to the exposed lysine residues on the ER, targeting it for degradation by the proteasome [4][13]. Additionally, fulvestrant has been shown to have a longer duration of action than other ER antagonists due to its ability to induce downregulation and degradation of the receptor, as opposed to simply blocking its activity [2][11].
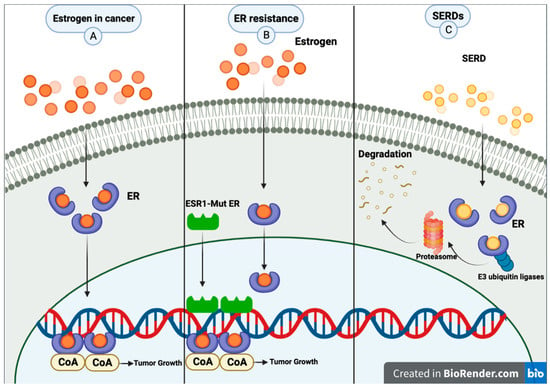
Figure 1. (A) Common pathway for estrogen in breast cancer. (B) Metastatic breast cancer patients may experience resistance mechanisms to endocrine therapy. A mutation of estrogen receptor 1 (ESR1) causes constant ER activity and enhanced transcription of ER-dependent genes without hormones, resulting in resistance to estrogen deprivation and aromatase inhibitor therapy. (C) SERDs bind to the estrogen receptor; then, E3 ubiquitin ligases and ubiquitinates the ER, marking it for degradation by the proteasome. The proteasome eventually degrades the ubiquitinated ER. Created with BioRender.com (accessed on 8 June 2023).
Overall, the unique molecular mechanism of the fulvestrant as a SERD has led to its successful use in treating advanced ER+ breast cancer, both as a first-line therapy and in patients who have progressed on prior endocrine therapies as a single agent or in combination with other ETs or targeted therapies [1][5][6][7][8][9,14,15,16,17].
However, fulvestrant has several limitations. Fulvestrant is only available as an injectable formulation, which can be inconvenient for patients who prefer oral medications or have difficulty with injections [9][18]. Compared to an AI, fulvestrant seems to be superior in treating ER+ metastatic breast cancer with an ESR1 mutation. ESR1 mutations play a crucial role in the development of endocrine therapy resistance in breast cancer. ESR mutations are expressed in the majority of breast cancers and are regulators of breast cancer development and progression. ESR1 mutations, particularly ones in the ligand binding domain, have accounted for acquired endocrine resistance in a significant fraction of patients with metastatic disease [10][19]. However, fulvestrant’s efficacy against ESR1 mutant metastatic breast cancer in the second line is modest, with median PFS ranging from 3 to 4 months [6][11][15,20]. Additionally and most importantly, after adopting CDK4/6 inhibitors in combination with AI as the standard first-line treatment for metastatic ER+ breast cancer a few years ago, newer clinical data have revealed minimal efficacy of fulvestrant monotherapy in the second line among these CDK4/6 inhibitor-treated tumors [12][21]. Several oral SERDs are being developed to address these drawbacks and are currently in clinical trials.
Furthermore, research indicates that fulvestrant-resistant breast cancer cells can proliferate independently of estrogen signaling and display downregulated estrogen signaling [13][14][22,23]. This information is crucial to guiding clinical application and future research in this area, as it provides insights into the drug’s mechanism of action and resistance patterns in breast cancer cells.
Studies show that fulvestrant’s primary anticancer effect is not through ER degradation but rather through the transient immobilization of ER receptors in the nucleus [15][24]. A SUMOylation process takes place in this action, leading to transcriptional disturbances and the elimination of receptors [16][17][18][25,26,27]. Studies have shown that fulvestrant induces transient binding of ERα to DNA, followed by rapid release without loss of nuclear localization. Unlike proteasome inhibition, this mechanism occurs before receptor degradation [16][25]. With fulvestrant, chromatin accessibility is reduced, suggesting that chromatin remodeling at ERα target regions prevents transcription despite receptor binding. The SUMO2/3 marks were also found on chromatin in cells treated with fulvestrant but not SERMs [16][19][25,28].
2. Camizestrant (AZD9833)
Camizestrant is a pure ER antagonist and next-generational oral SERD. SERENA 1 is a phase I, open-label, dose-dependent exposure trial that analyzed the safety, tolerability, and preliminary clinical efficacy of camizestrant monotherapy (part A (escalation)/part B (expansion)) and in combination with palbociclib (parts C/D), everolimus (E/F), abemaciclib (G/H), and capivasertib (I/J) in pretreated women with ER+ HER2− advanced breast cancer. In parts A/B, treatment-related AEs were visual disturbances, bradycardia, nausea, fatigue, dizziness, and vomiting. Three doses, 75 mg QD, 150 mg QD, and 300 mg QD, were proposed for a phase II study. One out of 7, 2 out of 11, and 2 out of 10 patients achieved ORR with these doses [20][39]. SERENA-1 parts C/D is an ongoing study with a multi-part open-label trial currently examining camizestrant combined with palbociclib. Data from camizestrant 75 mg QD in combination with the standard dose of palbociclib have been presented [21][40]. Some patients experienced bradycardia, GI disturbances, and visual disturbances likely related to camizestrant; however, most were grade 1. The clinical benefit rate at 24 weeks for these heavily pretreated patients was 28% with this combination [21][40]. This combination is further analyzed in ongoing phase III studies of SERENA-4 and SERENA-6 [21][40].
SERNA-2 is a randomized, open-label, multicenter phase II trial that analyzed the efficacy and safety of camizestrant with the various dosages of 75–300 mg administered as a monotherapy in women with ER+, HER2− previously treated advanced breast cancer in comparison with fulvestrant. The camizestrant 300 mg QD arm was discontinued after 20 patients were enrolled in that arm. Camizestrant significantly reduced the risk of disease progression by 42% at a 75 mg dose ((HR 0.58, 0.41–0.81); p = 0.0124; p = 0.0124; PFS of 7.2 vs. 3.7 months) and 33% at a 150 mg dose ((HR 0.67, 0.48–0.92); p = 0.0161; PFS of 7.7 vs. 3.7 months) compared to fulvestrant [22][41]. Among the ESR1 mutant tumors, median PFS with camizestrant 75 and 150 mg QD and fulvestrant were 6.3, 9.2, and 2.2 months, respectively, numerically favoring a benefit with camizestrant compared to fulvestrant, but this was not statistically significant as this phase II study was not powered to demonstrate benefit in the ESR1 mutant subset. Among the ESR1 wild-type tumors, the efficacy seemed similar numerically: 7.2, 5.8, and 7.2 months, respectively. Twelve percent of patients receiving 75 mg QD of camizestrant developed grade 3 or higher AEs compared to 13% receiving fulvestrant and 21% receiving camizestrant 150 mg QD. The most common AEs were bradycardia, visual disturbances, and fatigue.
Multiple ongoing trials are investigating camizestrant further. SERENA-3 is a randomized, open-label, parallel-group pre-surgical trial looking at different camizestrant 75–150 mg doses in postmenopausal and possible premenopausal women with ER+/HER2− primary breast cancer [23][42]. SERENA-4 is an ongoing randomized multicenter, double-blind phase III trial comparing the safety and efficacy of camizestrant plus palbociclib vs. anastrozole plus palbociclib in patients with ER+/HER2− previously untreated breast cancer [24][43].
SERENA-6 is a novel and exciting phase III clinical trial where patients with advanced ER+ HER2− breast cancer who are receiving an AI plus palbociclib or abemaciclib and have developed the ESR1 mutation but without overt disease progression will be randomized for a continuation of current treatment vs. switching AI to camizestrant with the continuation of the same CDK4/6 inhibitor [25][44]. The primary endpoint is PFS.
Oral SERDs have evolved into a significant advance in oncology when treating ER+ breast cancer. The agents in this class, exemplified by camizestrant (AZD9833), provide a means of overcoming the limitations associated with the first generation of SERDs, such as fulvestrant, which has a low bioavailability due to its intramuscular administration [26][45]. A phase III trial of oral SERDs has demonstrated efficacy in modulating ER-regulated gene expression and antiproliferation in ESR1 wild-type and mutant cells, demonstrating efficacy in treating early and advanced ER+ breast cancer. This addresses a critical need since ESR1 mutations are a major cause of endocrine therapy resistance. Oral SERDs are combined with CDK4/6 inhibitors and PI3K/AKT/mTOR-targeted therapy to enhance treatment efficacy and overcome resistance. However, despite these advances, there remain challenges associated with optimizing pharmacokinetics and understanding resistance mechanisms [27][28][46,47]. Future research is positioned to address these hurdles to improve patient outcomes in ER+ breast cancer treatment.
3. Giredestrant
Giredestrant is another nonsteroidal SERD designed to target ER+ breast cancer [29][48]. GO39932 is a 1a/1b multicenter, open-label, dose-escalation study investigating the safety profile and preliminary antitumor activity of giredestrant alone and giredestrant in combination with palbociclib. Participants have ER+ HER2− advanced or metastatic breast cancer [30][49].
In GO39932 cohort A, giredestrant as a single agent was well tolerated at all doses without any DLTs and with low-grade AEs (i.e., nausea, arthralgia, and fatigue), none of which required participants to discontinue treatment in phase 1a. It had encouraging antitumor effects and clinical benefits at all doses [31][50]. In GO39932 cohort B, a giredestrant with palbociclib was assessed. None of the participants had to discontinue treatment due to AEs. Adverse effects in >/= 10% of patients included neutropenia, fatigue, bradycardia, diarrhea/constipation, dizziness/nausea, and so forth; 57% of patients had >/= grade 3 AEs, and 50% of patients had >/= grade 3 neutropenia. No drug–drug interactions were observed between giredestrant and palbociclib [29][31][48,50]. The clinical activity reported for the 30 mg monotherapy arm and the 100 mg dose with the the palbociclib arm was encouraging, with ORR at 20.0% and 47.7%, respectively. The clinical benefit rates were 55.0% and 81.3%, respectively [32][51].
Next, acelERA BC is a randomized, open-label, multicenter phase 2 study. It investigated the safety and efficacy of giredestrant compared to the physicians’ choice, ET (fulvestrant or an AI), for patients with ER+ HER2− locally advanced or metastatic breast cancer who have already been treated with one to two types of systemic therapy [33][52]. The trial could not meet the primary endpoint of investigator-assessed PFS; nevertheless, the giredestrant did show numerical improvement compared to ET in ORR and clinical benefit rate. Additionally, in patients with the ESR1 mutation, their PFS was numerically higher with the giredestrant. Lastly, it was well tolerated and had a good safety profile consistent with others [34][53].
CoopERA is a completed randomized, open-label, multicenter phase 2 study investigating the efficacy, safety, and pharmacokinetics of giredestrant versus anastrozole within a 14-day window-of-opportunity phase followed by 16 weeks of a neoadjuvant treatment phase of giredestrant plus palbociclib vs. anastrozole plus palbociclib. The patient population is postmenopausal with ER+ HER2− untreated early breast cancer [35][54]. Giredestrant did indeed meet the primary endpoint and had higher Ki-67 suppression after week two compared to anastrozole. Note that this higher suppression continued up to surgery with giredestrant with palbociclib (−81% (95% CI: −86%, −75%)) versus anastrozole with palbociclib (−74% (95% CI: −80%, −67%)). Lastly, the safety profile is comparable to other giredestrant studies. This study shows that giredestrant demonstrates more antiproliferation in ER+ HER- early breast cancer than an aromatase inhibitor, anastrozole. Giredestrant with palbociclib also had a higher complete cell-cycle arrest at surgery, defined as Ki67 ≤ 2.7%, compared to anastrozole with palbociclib (20% vs. 14%) [36][55]. There are multiple ongoing studies investigating giredestrant in ER+ breast cancer where results are pending.
Also, evERA is an ongoing randomized, open-label, multicenter phase III study that investigates the efficacy and safety of giredestrant with everolimus versus exemestane with everolimus. Participants are patients with ER+ HER2− locally advanced or metastatic breast cancer [37][56].
Finally, lidERA is an ongoing randomized, open-label, multicenter phase III study investigating the efficacy and safety of giredestrant versus the physician’s choice, endocrine therapy. Participants are patients with medium-risk and high-risk histologically proven Stage I to Stage III confirmed ER+ HER2− early breast cancer [56,57]. Both pre- and postmenopausal women are eligible for this study.
4. Amcenestrant
Amcenestrant is an orally bioavailable SERD with pure ER antagonism in vivo [38][58]. AMEERA-1 is an open-label, single-arm study that evaluated amcenestrant monotherapy in postmenopausal women with ER+/HER2− advanced breast cancer who were heavily pretreated. In AMEERA-1 arm 1 part A-B, the optimal dosing of 400 mg QB of amcenestrant monotherapy was chosen for a phase 2 dose with no grade ≥3 TRAEs like cardiac/eye toxicities reported. The confirmed objective response rate was 5/46 (10.9%), with an overall clinical benefit rate (CBR) of 13/46 (28.3%). The wild-type ESR1 and mutated ESR1 showed similar CBRs of 34.6% and 21.1%, respectively [39][59]. The result showed that amcenestrant at RP2D of 400 mg QD monotherapy demonstrated antitumor activity regardless of baseline ESR1 mutation status and was well tolerated [39][59].
AMEERA-3 was a prospective, open-label, randomized phase 2 study to assess the safety and efficacy of amcenestrant compared to the ET of the physician’s choice in patients with ER+/HER2− metastatic or locally advanced breast cancer or metastatic breast cancer that progressed on ET. However, the trial did not meet its primary endpoint. The PFS was similar at 3.6 months for amcenestrant and 3.7 months for endocrine monotherapy [40][60]. TRAEs mainly were grade 1 or 2. In the I-SPY2 endocrine optimization protocol (EOP), the safety and efficacy of amcenestrant were evaluated with and without abemaciclib or letrozole [41][61]. The primary objective of the EOP is to assess the feasibility of treating molecularly selected patients with early stage ER+ HER2− molecular low-risk breast cancer with neoadjuvant endocrine therapy.
AMEERA-4 (NCT04191382) was a phase II preoperative window of opportunity that compared the safety and efficacy of two dose levels of amcenestrant and a standard dose of letrozole with paired biopsies assessed for biomarkers in a 1:1:1 randomization design among early stage ER+ HER2− breast cancer patients [42][62]. The primary endpoint was the change from baseline in Ki67 after two weeks of treatment with amcenestrant or letrozole using paired biopsies. The reduction in Ki67 was 75.9% (67.9, 81.9) for amcenestrant 400 mg, 68.2% (58.4, 75.7) for amcenestrant 200 mg, and 77.7% (70.0, 83.4) for letrozole. All TRAEs were grade 1 or 2 and were similar among three arms, ranging from 20 to 25%. The sponsors prematurely discontinued this trial due to their strategic decision to stop the development of this drug.
AMEERA-5, a phase III, randomized, double-blind, multinational study that analyzed women with ER+ HER2− metastatic breast cancer and compared amcenestrant plus palbociclib versus letrozole plus palbociclib failed to reach the prespecified boundary of continuation on interim analysis [43][63]. Based on this, the sponsor discontinued the global clinical development of amcenestrant.
5. Imlunestrant
Imlunestrant is another oral SERD in development. EMBER is an ongoing phase 1a/1b study with imlunestrant (doses 200–1200 mg) used as a monotherapy and combined with abemaciclib +/− an aromatase inhibitor (anastrozole, exemestane, or letrozole). In phase 1a, patients with ER+ HER2− advanced breast cancer with three or fewer prior therapies were recruited. Patients with ER+ endometrial cancer with prior platinum therapy were also recruited [44][64]. So far, monotherapy demonstrates no dose-dependent toxicities and TEAEs of mostly grade 1 or 2, including nausea, fatigue, and diarrhea. There was one grade 3 TEAE, diarrhea. Additionally, combination therapy (imlunestrant with abemaciclib +/− an aromatase inhibitor) in phase 1b has shown a satisfactory safety profile [45][65]. New findings presented at the 2022 San Antonio Breast Cancer Symposium regarding the combination therapy show favorable efficacy with a 12-month PFS compared to historical data from MONARCH 2 and 3 [46][66]. Another monotherapy trial is EMBER-2, an ongoing phase 1 study preoperative window studying the effects of imlunestrant on Stage I–III ER+ HER2− breast cancer in postmenopausal patients. As of yet, no results have been released [47][67].
EMBER-3 is an open-label, randomized 3-arm phase 3 study that compared the safety and efficacy of imlunestrant monotherapy vs. SOC ET (exemestane or fulvestrant) vs. imlunestrant plus abemaciclib among advanced ER+ HER2− breast cancer patients who have previously received an ET for advanced breast cancer [48][68]. EMBER-4 is another ongoing phase 3 study investigating imlunestrant compared to SOC ET in participants with high-risk ER+ HER2− early breast cancer. These participants will have already had two to five years of adjuvant endocrine therapy prior to this [49][69].
6. Rintodestrant
Rintodestrant is an oral SERD that competitively binds and degrades the estrogen receptor (ER). The safety and efficacy of rintodestrant were investigated in a phase 1 study, NCT03455270, among ER+ HER2− advanced breast cancer patients. Based on part 1 (dose escalation) and part 2 (dose expansion), the optimal dose of rintodestrant was 800 mg daily. Overall, 63% of patients developed TRAEs, and the most common TRAEs were hot flushes, fatigue, and nausea [50][70]. Part 3 of the study assessed the safety and efficacy of the combination of rintodestrant with palbociclib in patients with HR+/HER2− metastatic breast cancer that had already progressed on previous endocrine therapy. The overall CBR with this combination was 61% (61% among ESR1 wild type, 56% ESR1 mutant) [51][71].
7. AZD9496
AZD9496 is a preclinical compound that can be taken orally and is nonsteroidal, potent, and selective in the degradation and inhibition of ER activity [52][72]. Investigations of the effects of AZD9496, tamoxifen, and fulvestrant on estrogen-responsive genes in tumor samples were reported in a study. A human transcriptome array was used to measure mRNA levels in tumors treated with the drugs. Gene expression analysis was performed on tumors treated with the drugs. According to the study results, AZD9496 inhibits the expression of estrogen-responsive genes similarly to fulvestrant and tamoxifen. A dose-dependent inhibitory effect was also observed between fulvestrant and AZD9496 in MCF-7 cells compared to tamoxifen when estrogen-regulated genes were examined. A significant decrease in ESR1 mRNA levels was not observed in vitro, suggesting that the protein downregulation previously described is due to a decrease in protein levels rather than an increase in transcript levels [53][73].
A preclinical study showed that AZD9496 could antagonize and degrade the estrogen receptor in breast cancer cell lines, xenograft models, and patient-derived xenografts with mutations in the ESR1 gene. AZD9496 was more effective at inhibiting tumor growth when combined with the PI3K pathway inhibitors and a CDK4/6 inhibitor. Clinical trials of AZD9496 in phase I showed good tolerability and safety, and some patients who had been heavily pretreated could stabilize their disease for a prolonged period [53][73].
As part of a clinical trial conducted by Robertson et al. (NCT03236974), AZD9496 was compared with fulvestrant concerning the effects on changes in ER, progesterone receptors (PR), and Ki-67 biomarkers in patients who were newly diagnosed with ER+ HER2− breast cancer. A random allocation of patients was conducted between days 5 and 14 for treatment with AZD9496 and on day 1 for fulvestrant. Based on the study’s results, AZD9496 reduced the expression of ER, PR, and Ki-67, but the reductions were not superior to the fulvestrant ones. The plasma concentration of AZD9496 was lower than predicted, whereas the plasma concentration of fulvestrant was consistent with historical data. Neither treatment had significant safety concerns, and both were well tolerated [52][72].