Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Lara Saftić Martinović and Version 2 by Lindsay Dong.
Addiction is a complex brain disease influenced by genetic, environmental, and neurological factors. Psychostimulants, cocaine, and methamphetamine influence different cell types in different brain regions, with a focus on the neurons responsible for rewarding effects in the nucleus accumbens (NAc) and ventral tegmental area (VTA). Known markers for psychostimulant-induced neuronal plasticity in combination with droplet-based high-throughput single-cell sequencing divided the heterogeneity of cell populations in NAc and VTA into clusters, where all cells of the same type do not respond equally to exposure to psychostimulants.
- addiction
- psychostimulants
- psychostimulant-induced neuronal plasticity
1. Introduction
Behaviorally, addiction is defined as compulsive drug seeking and use despite negative consequences [1]. The underlying molecular mechanisms primarily involve the brain reward system, associated with dopamine release [2][3][4][2,3,4]. Acute drug exposure disrupts neurotransmitter signaling, localization, metabolism, and synthesis, resulting in neuroadaptive changes that contribute to the development of addiction [5][6][5,6]. Prolonged drug exposure induces neuroplastic changes, altering the structure and function of neurons and influencing synaptic connections, thus impacting reward neuronal circuitry and resulting in long-term behavioral changes [7][8][7,8].
Drugs of abuse are a diverse group of compounds classified into various categories based on their pharmacological effects and molecular mechanisms of action. Cocaine (COC) and METH are central nervous system stimulants that increase dopamine release in brain regions of the mesolimbic circuit, influencing reward and cognitive functions [9][10]. Although COC and METH have similar behavioral and physiological effects, there are some major differences. COC is quickly removed and completely metabolized in the body, while METH has a longer duration of action due to slower metabolism and remains unchanged in the brain longer than COC, leading to prolonged stimulant effects [10][11]. Both COC and METH are highly addictive stimulants that are widely recognized as one of the most abused drugs in the world [11][12][12,13]. Key brain regions involved in reward processing, influenced by COC and METH, include the nucleus accumbens (NAc) and ventral tegmental area (VTA). Long-term changes induced by psychostimulant exposure in these regions involve alterations in gene expression and cellular physiology [13][14][14,15]. Psychostimulant-induced synaptic plasticity is linked to the known mechanism of phosphorylation of extracellular signal-regulated kinases (ERK1/2) and cyclic adenosine 3′,5′-monophosphate (cAMP)-response element-binding protein (CREB), with cAMP-dependent protein kinase (PKA) also playing a role in drug-induced memory formation [15][16][17][18][16,17,18,19].
While some people only use drugs for experimentation and never develop an addiction, others develop an addiction after being exposed to psychoactive substances on a regular basis. Understanding the molecular basis for this phenomenon requires a complex interplay of genetic, environmental, and neurobiological factors. Individual variability and the dynamic nature of drug responses both influence the molecular mechanisms involved in the cascade of events that affect neurotransmitter release, receptor activation, and intracellular signaling pathways. Individual vulnerability and susceptibility to addiction were studied in a mouse model, and it was discovered that in wild-type populations, voluntary oral methamphetamine consumption can undergo bidirectional selective breeding, producing two strings with high and low preference for METH [19][20]. Using a selective breeding approach, candidate genes can be identified in the absence of METH exposure. By using RNA-Seq to selectively breed low- and high-METH-preference mice, it was discovered that the trace amine-associated receptor 1 (Taar1) gene plays an important role [20][21].
Previous studies on molecular changes associated with addiction have primarily focused on candidate genes identified by proteomic or genetic techniques using tissue extracts from drug-exposed individuals. With this approach, it is not possible to distinguish in which cells the genetic changes originate, whether they occur at the gene or protein level and whether the observed changes are time-dependent or stable. To explain the complexities of these influences, it is necessary to refine the accepted central dogma, foundational to our understanding of genetic information flow, by applying epigenetics and epitranscriptomics to gene regulation (Figure 1).
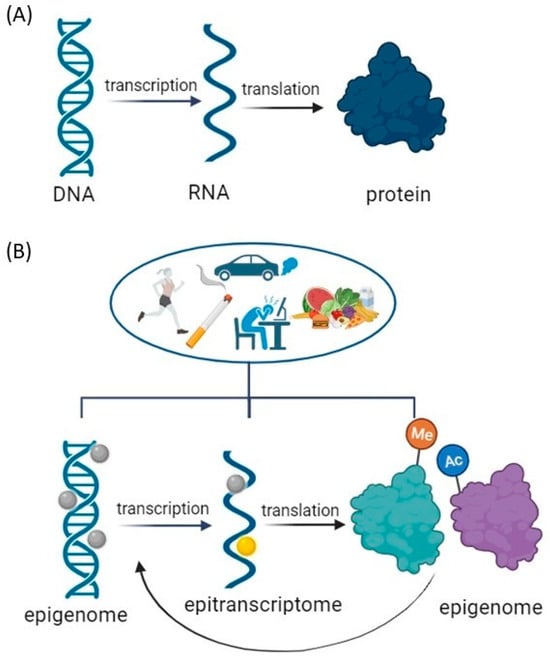
Figure 1. Modifications to the Central Dogma of Molecular Biology: Gene Regulation Modulation by Environmental Factors. (A) Central Dogma: a theory stating that genetic information flows only in one direction, from DNA to RNA to protein, or RNA directly to protein. (B) Environmental Modulation of Central Dogma: Impact on DNA Unfolding, Transcription, and Translation Processes. Epigenome modification = DNA methylation or acetylation and post-translational modifications of histone tails that include phosphorylation, ubiquitination, acetylation, and methylation. Epitranscriptome modifications = RNA modifications primary messenger RNA (mRNA) modifications: N1-methyladenosine (m1A), N6–methyladenosine (m6A), 5–methylcytosine (m5C), pseudouridine (Ψ), and others. Created with BioRender.com (accessed on 11 December 2023).
2. Single-Cell Sequencing Techniques
Traditional classification divides cell types based on morphology rather than molecular features [21][22][31,32]. Despite the fact that cells have almost identical genotypes, traditional RNA sequencing gives an average expression profile from a population of cells, ignoring cell-to-cell variability and transcriptome information from a subset of active genes [23][24][25][33,34,35]. Single-cell RNA sequencing (scRNA-seq) is used for genetic profiling of cells that require prior separation of one cell type from a cell suspension using a fluorescence-activated cell sorting and flow cytometer [26][36]. This approach requires a priori knowledge of a cell-type-specific marker, which is dependent on the availability of appropriate antibodies [27][37]. Great progress in scRNA-seq was made by applying high-throughput droplet-based techniques, which do not require prior separation of the cells from the cell suspension, namely, InDrop [28][38], Drop-seq [29][39], and 10× Chromium [30][40], supporting cost-effective capture and library production for thousands to millions of cells and allowing examination of gene expression heterogeneity among individual cells (Figure 2).
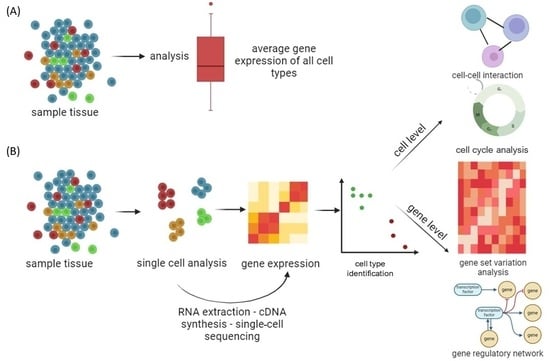
Figure 2. Genomics, transcriptomics, and epigenomics technologies focused on the characterization of individual cells from tissue samples. (A) Conventional massive population sequencing provides average expression signals for different cells, ignoring cell-to-cell variability. (B) By applying high-throughput droplet-based scRNA-seq to complex cell populations, it is possible to uncover different cell types and interactions between cells, follow the development of distinct cell lineages, track changes in gene expression, and identify regulatory relationships between genes. Created with BioRender.com (accessed on 11 December 2023).
2.1. Application of Single-Cell Sequencing in Addiction Research
By defining region-specific molecular signatures and neuronal circuits involved in addictive behaviors [31][32][33][48,49,50], droplet-based high-throughput scRNA-seq enabled researchers to compare gene expression profiles in different brain regions involved in addiction [34][51]. At the molecular level, scRNA-seq of individual neurons identifies specific neuronal subtypes associated with reward, reinforcement, and addiction processes [35][52], whereas transcriptome analysis identifies distinct transcriptional signatures associated with addiction-related behaviors [36][53]. Understanding how individual cells within a population exhibit different sensitivities or adaptations to drug use is crucial, while interactions between the nervous and immune systems must be integrated in this aspect.
2.1.1. Identifying Cell Populations by Molecular Clustering
scRNA-seq has been shown to be an effective tool for characterizing cellular diversity in brain regions associated with the reward system, such as the NAc and VTA [37][38][54,55]. The NAc is a brain region primarily composed of two main types of cells, namely, Medium Spiny Neurons (MSNs) and interneurons. In NAc, clustering analysis revealed nine major cell populations, four neuronal and five non-neuronal [37][54]. Using this approach, in the NAc, novel subpopulations of interneurons and MSNs were identified [35][52]. scATAC-seq has been used to map cell-type-specific differences in chromatin accessibility in the NAc, providing insights into the epigenomic landscape of this brain region [38][55]. Moreover, the characterization of γ-Aminobutyric Acid (GABA) MSNs and the discovery of notable variations in receptor expression patterns and MSN activation within NAc subterritories have highlighted the anatomical and functional heterogeneity of the NAc [39][56]. The ventral tegmental area (VTA) is best known for containing dopaminergic neurons associated with reward and motivation. scRNA-seq identified in VTA selective markers for dopamine and combinatorial neurons revealed expression profiles for drugs of abuse receptors and population-specific enrichment of genes associated with brain disorders [40][57]. This comprehensive molecular characterization highlights the heterogeneity of the NAc and VTA cell population, providing a valuable resource for future research into VTA and NAc gene expression and its implications for reward-related behaviors such as addiction.
By applying scRNAseq in the study of acute COC administration in rodents, distinct neuronal clusters within the NAc were discovered. These clusters exhibited known markers associated with two main types of MSNs: dopamine receptor D1–positive (Drd1-MSNs) and dopamine receptor D2–positive (Drd2-MSNs) [41][58]. The same neuronal substrate that allows drugs of abuse to access Drd1 and Drd2 MSNs in NAc has been confirmed to augment and corrupt a shared pathway that normally serves physiological needs [42][59].
METH is another commonly abused psychostimulant that induces synaptic plasticity and pathological memory enhancement [43][61]. Epigenetics plays an important role in regulating METH addiction [44][62]. METH modulates dopamine (DA), norepinephrine (NE), serotonin, glutamate (Glu), and GABA neurotransmitters in the medial prefrontal cortex (mPFC), the VTA, and the NAc through histone acetylation, methylation, micro RNAs (miRNAs), and ubiquitination. These epigenetic mechanisms do not regulate METH-induced addiction alone but rather collaborate with miRNA regulation of the Ubiquitin proteasome system [44][62]. Cell clustering was performed on bulk RNA data using single-cell RNA data extracted from astrocytes for analysis of differently expressed genes in METH-exposed mice. The NF-κB signaling pathway, inflammation, and neurodegeneration were among the considerably enriched pathways found in the analysis. Immune infiltration analysis showed that the METH group had significantly lower neutrophil infiltration and significantly higher monocyte, T, and NK cell infiltration [45][63]. Strong inflammatory responses occur early in METH withdrawal, and they decrease as withdrawal time increases.
2.1.2. Identifying Transcriptome Mechanism
By analyzing single-cell transcriptional responses in the prefrontal cortex (PFC) cells of mice undergoing COC self-administration, specific cell types that express genes associated with COC addiction were identified, namely, ∆FosB, Methyl CpG binding protein 2 (MeCP2) and brain-derived neurotrophic factor (BDNF) [46][68]. Following COC administration, ∆FosB was reported to also be expressed in the VTA with dopamine neurons projecting to the NAc [47][69], MeCP2 was reported to be altered in the NAc [48][70], and BDNF expression played a role in the VTA-NAc pathway [49][71]. These genes were selectively expressed in excitatory neurons, inhibitory neurons, and non-neuronal glial cells while showing varied expression levels over the stages of COC addiction [46][68]. Reactive oxygen species and oxidative stress in striatal regions are known to be elevated by COC, and this effect is further compounded by elevated glutamate release and excessive dopamine levels. The potential involvement of both cell types in the regulation of conserved gene networks was revealed by integrating human transcriptomes with the Drd1 and Drd2 MSN transcriptome data from mice. Numerous transcription factors that are predicted to be upstream of the abnormalities in gene expression linked to addiction were identified through this analysis. The majority are shared between the NAc and dorsum striatum. Notably, several of these predicted upstream transcription factors have been implicated previously in COC addiction in rodent models; in particular, Activator protein 1 (AP-1) (FOS/JUN) family, EGR family, NFκB, E2F, and several nuclear hormone receptors cells [50][72]. In response to acute and chronic METH, mice hippocampi exhibit significant volumetric atrophy compared to controls. The genes involved in cytoskeleton organization and phagocytosis were downregulated in the acute METH-treated group compared to the control group. In the group receiving long-term METH treatment, genes linked to synaptic transmission, neuron differentiation regulation, and neurogenesis regulation were downregulated [51][73]. Drd1 overstimulation after METH exposure induces metabolic changes and transcriptional pathways, switching gene expression and neuronal phenotype underlying addictive behavior [52][74]. PKA phosphorylates voltage-dependent ion channels, GLUT receptors, transcription factors, and epigenetic enzymes involved in synaptic plasticity as naturally occurring in normal striatum. When the Drd1 is activated, PKA activates mitogen-activated protein kinases (MAPKs) and extracellular signal-regulated kinases 1/2 (ERK1/2) [53][75]. Nuclear receptors, CREB, Elk-1, and H3 histones are all phosphorylated by ERK1/2 upon translocation to the nucleus, where they control the expression of certain genes [54][76]. The DA- and cAMP-regulated phosphoprotein (DARPP-32) is a key substrate of DRD1/PKA signaling in the striatum [55][77]. METH consumption results in oxidative stress in the DA terminals due to excess of free DA undergoing oxidative metabolism and autoxidation, intracellularly and extracellularly. Along with hydrogen peroxide and reactive oxygen species (ROS) production, toxic DA metabolites like quinones and 3,4-Dihydroxyphenylacetaldehyde promote structural modifications of proteins. METH influences other cell organelles, endoplasmic reticulum, and mitochondria, leading to neurotoxic effects, while ROS accumulation leads to misfolded and insoluble proteins and severed organelles, decomposed by cell-clearing mechanisms of autophagy and ubiquitin proteasome [56][78].3. Epigenetic Mechanisms and Psychostimulant Addiction
3.1. Cocaine-induced Epigenetic Modifications
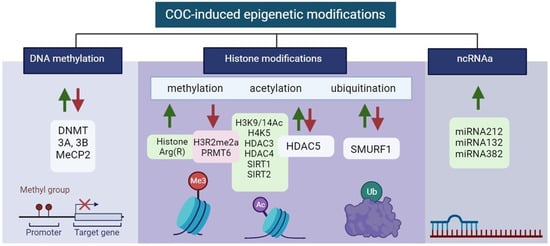
Epigenetic modifications in the control of gene expression of specific brain regions in COC addiction are summarized in Figure 3.Figure 3. Illustration of aberrant alterations in COC-induced epigenetic modifications. Green and red arrows indicate upregulation and downregulation, respectively. Created with BioRender.com (accessed on 13 December 2023).
3.1.1. DNA Methylation
Environmental stimuli in COC exposure can be translated to changes in gene expression and phenotypes that, through enzymatic modifications to DNA sequences, create long-lasting behavioral phenotypes. These methylated DNA regions are bound by methyl-binding domain-containing proteins such as methyl CpG-binding protein 2 (MeCP2), which is important for recruiting co-repressors such as methyltransferases to the gene promoter [57][100]. DNA methylation is critical for imprinting, X chromosome inactivation, and cell differentiation. Therefore, it is important that it can be altered at specific loci in germ cells by exposure to environmental factors such as toxins [58][101] and stress [59][102], and can therefore be inherited by offspring over multiple generations. Acute cocaine administration increases DNA methylation and DNMT3A and DNMT3B levels, while the binding of MeCP2 to specific gene promoters can decrease gene expression in the NAc [60][103].3.1.2. Histone Modifications
Histones Methylation
Histones H3 and H4 can have their lysine or arginine residues methylated, with varying effects on transcription. While lysine methylation is connected to both transcriptional activation and repression depending on the methylation site, arginine methylation encourages transcriptional activation [61][106]. This adaptability could be explained by the fact that, in contrast to acetylation, methylation has no effect on histone charge or histone–DNA interactions directly. H3 methylation increases the gene transcription, while lysine (K) methylation on H3K9Me3 and H3K27Me3 at specific gene regions represses transcription [62][107].Histones Acetylation
COC administration leads to significant acetylation of histones H3 and H4 [63][109]. H3K9, H3K14, H4K5, H4K8, H4K12, and H4K16 have emerged as the most extensively researched acetylation sites in the context of COC exposure [64][90]. Histone acetylation in the NAc is associated with CREB-binding protein (CBP), a histone acetyltransferase [65][110]. Repeated exposure to COC leads to a dual effect on histone acetylation, resulting in H3Ac/H4Ac-increased and -decreased acetylation of gene promoters, attributed to the hypoacetylation of histones H3 and H4 [63][109]. In the VTA during COC withdrawal, there is an observed enrichment of CBP in the promoter region of BDNF leading to an elevated level of histone acetylation, particularly H3K9/14Ac, at the Bdnf promoter [66][111].Histones Ubiquitination
The ubiquitin-proteasome system (UPS), one of the epigenetic hallmarks, is a multifaceted network of ubiquitin ligases and proteasome structures that controls synaptic and epigenetic plasticity [67][121] and is also involved in memory processes and substance use disorders [68][122]. E3 ubiquitin-protein ligase SMURF1 is a key mediator of neuroadaptations in the nucleus accumbens that follow COC exposure and mediates cue-induced COC seeking during withdrawal [69][123]. All of these results point to the possibility that the SMURF1–SMAD1/5–RUNX2 pathway functions as a crucial transcriptional regulator that modifies plasticity after COC self-administration. The finding that RUNX2 and SMAD1/5 are upregulated in the NAc following COC self-administration allowed researchers to investigate RUNX2 binding to target genes that had previously been connected to COC plasticity. RUNX2 is found to bind at AP-1 sites and to the promoters of these genes following COC self-administration, indicating that it may be a master regulator of several pathways regulating COC-induced plasticity [70][124]. There is a critical need for new research to identify novel potential therapeutic targets in order to develop effective treatments for COC use disorder.3.1.3. ncRNAs
The most commonly investigated ncRNAs involved in epigenome modifications in COC addiction are miRNAs. They can regulate COC intake and potentially have an impact on developing compulsive consumption of the drug. The mechanisms that stand by miR-212 influences COC intake were also a point of exploration. The cAMP signaling cascade strongly activates the miR-212/132 gene cluster, with CREB upregulating both miRNAs. It was found that the pathway of miR-212 regulation dramatically boosts CREB signaling in cultured cells and also in the striatum of rats [71][104]. miR-206 has been shown to negatively regulate BDNF expression, which is known to be important for the motivational effects of COC and other addictive substances [72][126].3.2. Methamphetamine-Induced Epigenetic Modifications
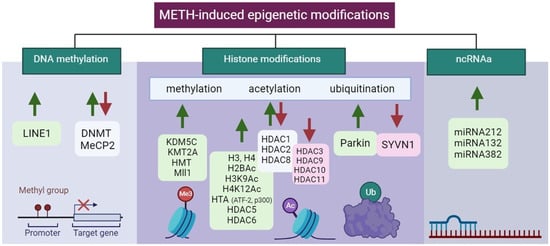
Epigenetic modifications in the control of gene expression of specific brain regions in METH addiction are summarized in Table 2 and Figure 4.Figure 4. Illustration of aberrant alterations in METH-induced epigenetic modifications. Green and red arrows indicate upregulation and downregulation, respectively. Created with BioRender.com (accessed on 13 December 2023).
3.2.1. DNA Methylation
DNA methylation levels have been found to be changed in METH addicts [73][129], and moreover, this also occurs in their offspring [74][130]. BDNF methylation was increased in the PFC of METH-addicted rats and patients but was decreased in the hippocampus of rats [75][131]. The neurotoxic effects of METH exposure were partly caused by a decrease in BDNF expression [75][76][131,132].