Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Carlo De Salvo and Version 2 by Catherine Yang.
Gastric cancer is one of the leading causes of cancer deaths worldwide, with chronic gastritis representing the main predisposing factor initiating the cascade of events leading to metaplasia and eventually progressing to cancer. Th2 immune responses play a major role in the events causing chronic inflammation leading to tumorigenesis.
- gastric metaplasia
- spasmolytic polypeptide-expressing metaplasia (SPEM)
- intestinal metaplasia
- Th2 immunity
1. Th2 Immune Responses, Specifically via IL-13, Is Central to the Pathogenesis of Gastric Cancer
In H. pylori-related inflammatory responses, a predominant Th1 response is present in the majority of H. pylori-positive patients [1][2][90,91]. However, a Th1 to Th2 shift has been observed in patients with gastric cancer and dysplasia [3][29]. In fact, a mixed Th1/Th2 immune response is associated with non-atrophic chronic gastritis, while a prevalent Th2 immune response, mostly IL-13-mediated, characterizes patients with intestinal metaplasia and intestinal-type gastric cancer [4][38]. While a Th1-mediated immune response is considered mostly proinflammatory, Th2 responses can be immunoregulatory and protective and have the ability to promote antibody-mediated immunity against extracellular parasites and bacterial infection [5][6][92,93]. In fact, the overexpression of IL-1β in the gastric epithelium can promote gastric inflammation, atrophy, and metaplasia [7][94], and the knocking out of IL-1 has been shown to be protective against pathologic processes induced by H. pylori PMSS1 [8][76]. Similarly, the transgenic expression of interferon (IFN)γ, stromal-derived factor (SDF)-1, or IL-8 (the latter in transgenic mice bearing a human IL-8 bacterial artificial chromosome), accelerates gastritis and metaplasia during Helicobacter infection [9][10][11][95,96,97]. Conversely, the administration of IL-33 triggers a Th2 immune response, promoting metaplasia in the stomach. In fact, IL-33 is required to induce metaplasia after acute parietal cell loss during the development of SPEM by promoting a Th2 inflammatory response, mainly mediated by IL-13, and the recruitment of eosinophils [12][25] and macrophages M2 [13][14][69,98].
A seminal study in 2018 revealed that Th2, and notably not Th1 or Th17, immune responses are responsible for mediating the link between gastritis and gastric cancer; specifically, CTLA4 deficiency induces Th2-dependent immune dysregulation that results in the development of gastric cancer [15][99]. More recently, Noto et al. identified six cell subtypes (i.e., mast cells, CD19+ B cells, macrophages, group 2 innate lymphoid cells (ILC2s), as well as CD4+ and CD8+ T cells) as the main source of IL-13 in the context of gastritis, with mast cells being the greatest producers. In this study, the IL-13 receptor, an IL4Rα-containing heterodimer, is expressed on gastric epithelial cells, and IL-13 is crucial for the expansion of neck cells and subsequent SPEM by directly acting on epithelial cells. Indeed, IL4Rα-deficient mice develop gastritis, with immune cell infiltration and protein production resembling that found in wild-type controls but with no progression to SPEM, revealing the importance of IL-13 signaling during gastric carcinogenesis [14][98]. In line with these findings, a recent study identifying AQP5 in SPEM glands and at the base of incomplete intestinal metaplasia glands demonstrates that AQP5 expression is under the control of IL-13, further reinforcing the concept that this cytokine plays a fundamental role in the development of metaplasia and possibly, its progression to cancer [16][64]. Another recent study identified TNF+ Tregs as a source of IL-13 and proposes that IL-13 promotes self-renewal, migration and colony formation of gastric cancer cells via the phosphorylation of STAT3 [17][100].
2. IL-33 Is a Crucial Inducer of Th2 Responses Primarily, but Not Exclusively, by Inducing IL-13
IL-33 is widely distributed throughout various organ systems, both in non-hematopoietic and hematopoietic cells, the latter particularly in restricted populations of professional antigen-presenting cells, such as macrophages [18][19][101,102]. IL-33 was initially associated with Th2 immunity, based on the expression of its cell-bound receptor, ST2L, on polarized Th2 lymphocytes [18][101] and more recently, on ILC2s [20][103], and its ability to effectively induce M1, but more commonly M2, macrophage differentiation [21][104]. Furthermore, IL-33 is also considered a master regulator for eosinophil activation and recruitment into the mucosa of gastrointestinal and respiratory tracts interacting with the external environment [18][22][23][101,105,106]. High serum IL-33 concentration is reported to be associated with a poor prognosis in gastric cancer patients [24][107], and in vitro experiments suggest that IL-33 confers chemo-resistance [25][108] and increases invasiveness [26][109] to gastric cancer cells.
While the importance of IL-13 in gastric metaplasia seems to be mostly related to mucus hypersecretion [27][28][110,111], which is a specific feature of SPEM [29][30][87,112], IL-33-dependent induction of IL-13 is required to promote metaplasia following parietal cell loss and chief cell transdifferentiation into a mucus-producing cell type. It has been recently observed that the loss of GRIM-19, a mitochondrial gene commonly downregulated during gastric carcinogenesis, promotes SPEM in an IL-33-dependent manner; mechanistically, GRIM-19 deficiency, through the upregulation of NK-kB, leads to the activation of the NLRP3 inflammasome, which in turn, induces IL-33 expression and release [31][113]. Interestingly, IL-33 receptor-deficient mice do not develop SPEM following L635 treatment; however, the administration of recombinant IL-13 to these mice restores SPEM development, suggesting a major role of IL-13 in the pathogenesis of SPEM and for IL-33 as its upstream inducer [13][69]. In addition, IL-33-driven M2 macrophage polarization in L635-treated mice is associated with progression to a more advanced metaplasia, although not required for metaplasia induction [13][32][68,69]. Recently, epithelial-derived WAP four-disulfide core domain 2 WFDC2 was shown to induce SPEM via an IL-33-dependent promotion of M2 macrophage polarization; specifically, using chemically induced gastritis mouse models, the absence of Wfdc2 results in reduced M2 infiltrates and no progression to SPEM and dysplasia, while the administration of rWFDC2 or recombinant IL-33 restores the development of SPEM [33][114].
Furthermore, metaplasia is thought to serve a regenerative function in response to tissue damage and appears to facilitate epithelial repair [30][112]. In line with this concept, the IL-33/ST2 axis plays a critical role in gut mucosal wound healing by promoting epithelial repair and restitution, overall restoration of barrier integrity, and the resolution of inflammation. IL-33 can initially sustain inflammation in the colon of mice immediately after acute dextran sodium sulfate (DSS) challenge; however, its primary role is to promote mucosal wound healing during recovery [34][115]. As such, while the IL-33/IL-13 axis may be important in promoting pro-tumorigenic processes, it can also conceivably be critical for gastric epithelial repair. These findings are consistent with previous studies that associate decreased IL-33 in TFF2-deficient mice with defective metaplasia development and delayed downstream gastric epithelial repair [30][35][36][112,116,117]. Moreover, mice deficient for IL-33, ST2 and IL-13, after acute treatment with L635, show diffuse parietal cell loss and reduction in the zymogen granule maturation transcription factor Mist1, but chief cells fail to undergo complete differentiation [13][69], indicating the need for an appropriate cytokine response to promote transdifferentiation as part of the repair processes for the gastric epithelium. In fact, IL-33 has been proposed to function as an autocrine stimulus for gastric carcinogenesis. IL-33 and its receptor ST2 are found to co-localize in poorly differentiated human gastric cells; functionally, IL-33 self-stimulation promotes the survival and proliferation of cancer stem cells, supposedly in cooperation with the Wnt-axis, and confers resistance to chemotherapy [37][118].
3. Cell Types Associated with the Cascade of Events Leading to Gastritis, and Subsequently to Gastric Metaplasia
IL-33, in fact, has emerged as an essential mediator for the development of eosinophil-mediated allergic inflammation and asthma [22][23][105,106] and plays a pivotal role in eosinophil recruitment and helminth expulsion after parasitic hook worm infection [23][106]. The effects of IL-33 on the gastric immune compartment include massive infiltration of both eosinophils and M2 macrophages [13][69], further perpetuating a chronic inflammatory state. Importantly, the influx of eosinophils during gastric metaplasia and cancer development is thought to have both pathogenic as well as protective functions. A prior study investigating early gastric cancers showed the presence of tumor stromal eosinophils with morphologic evidence of activation, as well as the presence of tumor cells in intimate contact with activated eosinophils that results in focal cytopathic changes [38][119]. Eosinophil development and recruitment depend on IL-5, which represents the primary factor for eosinophil maturation and differentiation but is also important for their activation and recruitment. Chemokine ligand (CCL) 11 and CCL24, also known as eotaxin-1 and eotaxin-2, respectively, are eosinophil-specific chemokines that bind to the chemokine receptor, CCR3, expressed on the surface of eosinophils, and are critical for eosinophil recruitment [39][40][120,121]. Eosinophils are associated with chronic intestinal inflammation, and IL-33 induces eosinophil infiltration into the gut and a potent mucosal Th2 immune response [41][122]. In the stomach, eosinophils are present in inflammatory infiltrates within the gastric mucosa during Helicobacter infection [42][43][123,124]. Moreover, IL-33 activation and recruitment of eosinophils into the gastric mucosa during chronic inflammation is central for initiating the cascade of events promoting SPEM [12][25]. Eosinophils play an important role during the early events leading to gastritis/metaplasia; however, there are other cell types belonging to the innate immune system that are responsive to IL-33 and may be involved in the gastritis/metaplasia process, such as IL-33-activated M2 macrophages, that in turn are potent producers of IL-33. In fact, it is well established that IL-33 has the ability to increase overall macrophage numbers in the stomach and to polarize them to an alternatively activated M2 phenotype, albeit in the periphery (peritoneum) [36][117]. Mice lacking IL-33 or ST2 with advanced SPEM have markedly decreased numbers of M2 macrophages, but administering rIL-13, which acts downstream of the IL-33/ST2 axis, can revert this condition in ST2-deficient mice [13][69]. In the SAMP model of chronic gastritis, eosinophils appear to be the main cell type driving M2 macrophage activation, and depleting eosinophils has a similar effect as blocking IL-33 in regard to reducing gastric M2 macrophage activity, which in turn, can also release IL-33, resulting in a vicious cycle that fuels itself. [12][25]. On the other hand, eosinophils do not seem to be relevant in the L635-induced model of SPEM, which, differently from the spontaneous chronic model of inflammation, develops SPEM via chemically induced parietal cell obliteration. Such differences in the model used can also account for the different cell types mainly involved in the initiation and perpetuation of advanced SPEM [13][69].
It has been recently shown that IL-33-activated mast cells, together with tumor-associated macrophages, allow for the progression of tumor angiogenesis and are correlated with poor survival in gastric cancer patients [44][125]. Mast cells and eosinophils interact in a complex self-perpetuating cycle, wherein eosinophils produce mediators stimulating mast cell differentiation, activation, proliferation and survival [45][126]. In addition, activated mast cells release IL-5 and granulocyte–monocyte colony-stimulating factor that induce eosinophil recruitment and activation [46][127]. Furthermore, IL-33 stimulation, in vitro, induces mast cells to release IL-2, which, in turn, promotes the differentiation and expansion of ICOS+ Tregs that inhibit CD8+ T cells. Notably, in a xenograft gastric cancer model adopting NOD/SCID mice, this IL-33/IL-2-dependent activation of ICOS+ Tregs is shown to promote tumor growth and progression [47][128].
Finally, a subtype of the ILC family, ILC2s, are regulated by the transcription factor GATA3 [48][129] and are stimulated by IL-33, IL-25, and TSLP to provide an innate source of type 2 cytokines [49][50][130,131]. ILC2s are involved in tissue remodeling, mucus metaplasia, eosinophilia, and alternative macrophage activation through the production of type 2 cytokines [51][132]. The role of ILC2s in promoting SPEM is controversial. ILC2s are reported to be indispensable for the induction of SPEM following injury-mediated loss of gastric parietal cells, where they accumulate in an IL-33-dependent fashion. Specifically, ILC2s-dependent tissue repair processes of the gastric mucosa after injury entail a series of changes in both mesenchymal and hematopoietic cell compartments, which go through chief cell reprogramming, tuft cell and mucin-secreting foveolar cell proliferation, as well as immune cell recruitment [52][133]. In this same study, gastric ILC2s exhibit a specific, metaplasia-associated transcriptomic profile associated with the development of SPEM [52][133]. Conversely, in chronic gastritis-prone SAMP mice, the increase in ILC2 frequency is quite modest, and the precise contribution of ILC2s to the SAMP gastric phenotype remains unknown. In fact, SAMP mice lacking T/B cells but with intact ILC function (i.e., SAMP x Rag2−/− strain) have virtually normal stomachs, suggesting that the role of ILCs is negligible for SPEM development, perhaps because of their inability to mount adaptive immune responses and sustain a chronic inflammatory state [12][25].
Nonetheless, it has been reported that IL-33 induces IL-13 production from ILC2s to promote intestinal goblet cell differentiation within the colons of mice [53][134], indicating that the IL-33/IL-13 axis might play a relevant role in inducing intestinal metaplasia in the context of chronic gastritis. Since IL-33 is an alarmin released by damaged epithelial cells and activated macrophages in the context of Th2 responses, it accumulates in the inflamed gastric mucosa, where it activates ILC2s to produce IL-13 [54][135]. In a recent paper, O’Keefe et al. showed that ILC2-derived IL-13 stimulates the expansion of tuft cells and induces them to produce IL-25, which in turn further promotes the activity of ILC2s. This feed-forward circuit is thought to facilitate both SPEM development and its subsequent transition to gastric cancer. Indeed, tumor-bearing gp130−/− mice, when treated with pharmacological inhibitors of IL-13 or IL-25, are shown to bear smaller tumors containing fewer ILC2 and tuft cells. It is also suggested that, in patients with intestinal-like gastric cancer, a higher expression of gene signatures for ILC2s and tuft cells strongly correlates with reduced survival, further reinforcing the notion that this Th2 circuit contributes to the development and progression of gastric cancer, [55][136]. Furthermore, recent findings indicate a higher density of intratumoral IL-25-expressing macrophages that positively correlates with overall patient survival, possibly by regulating the T effector/T reg ratio [56][137] (Figure 12).
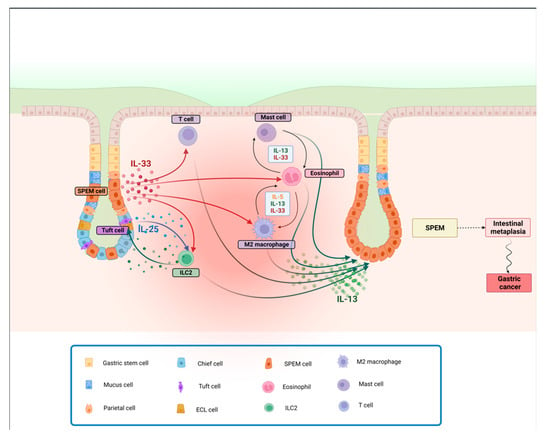
Figure 12. Th2-depenent gastric metaplasia onset and progression to SPEM. Following parietal cell loss, IL-33 is released, promoting activation and recruitment of different cell types involved in Th2 immune responses. IL-25, released by Tuft cells, also contributes, along with IL-33, to potentially induce the activation and proliferation of ILC2s. M2 macrophages and eosinophils, in turn, produce IL-13 and more IL-33, self-sustaining a feed-forward cycle, stimulating mast cell activity. The resulting downstream release of IL-13 promotes the progression to SPEM, that, in the presence of a chronic inflammatory insult, can lead to intestinal metaplasia and cancer. Abbreviations: IL, interleukin; SPEM, spasmolytic polypeptide-expressing metaplasia.