Anderson–Fabry disease (AFD) is a lysosome storage disorder resulting from an X-linked inheritance of a mutation in the galactosidase A (GLA) gene encoding for the enzyme alpha-galactosidase A (α-GAL A). This mutation results in a deficiency or absence of α-GAL A activity, with a progressive intracellular deposition of glycosphingolipids leading to organ dysfunction and failure. Cardiac damage starts early in life, often occurring sub-clinically before overt cardiac symptoms. Left ventricular hypertrophy represents a common cardiac manifestation, albeit conduction system impairment, arrhythmias, and valvular abnormalities may also characterize AFD. Even in consideration of pleiotropic manifestation, diagnosis is often challenging. Thus, knowledge of cardiac and extracardiac diagnostic “red flags” is needed to guide a timely diagnosis.
- Anderson–Fabry disease
- cardiomyopathy
- cardiac involvement
1. Introduction
2. General Features and Clinical Presentation of AFD
An AFD prevalence of 1/40,000–1/117,000 has been reported [2[2][9],9], although it appears to be underestimated [10,11,12,13][10][11][12][13]. Furthermore, a ten-year screening on 2034 probands with clinically suspected AFD significantly improved the rate of confirmed diagnosis [14], detecting 1.8% of GLA mutations [14]. The spectrum of disease severity is linked to the activity level of α-Gal A, which can range from deficiency to complete absence. A severe reduction in α-Gal A activity (<1% of mean normal) is associated with the classic form of AFD in hemizygous males, which is characterized by early clinical presentation, multiorgan involvement, more severe clinical manifestations, and adverse prognosis [6,15,16,17][6][15][16][17]. Males with a higher level of α-Gal A activity exhibit late-onset AFD with less severe disease expression than in the classic form [15,18][15][18]. The cardiac variant has also been identified within the non-classic forms, in which cardiac manifestation may be the exclusive or predominant disease expression. In these patients, differential diagnosis from other causes of LVH, such as cardiac amyloidosis (CA), hypertrophic cardiomyopathy (HCM), or hypertensive heart disease (HHD) is more challenging [19,20][19][20]. Since AFD follows an X-linked pattern of inheritance, affected females present with a slightly reduced to a near normal level of α-Gal A activity with the result that disease manifestation and prognosis are less severe than in their male counterparts. However, severe disease may also occur in females, who resemble the classic male phenotype of AFD patients [21]. Indeed, X-chromosome random inactivation (lyonization) leads to some cells expressing the normal allele and others with a mutated allele [21]. This leads to pleiotropic manifestations ranging from mild to severe disease expression. Beyond the inheritance pattern and α-Gal A mutation, other factors such as genetic modifiers, environmental factors, and epigenetics may impact disease spectrum manifestation. These factors also contribute to inter and intra-familial variation [6]. The heterogeneous clinical presentation of AFD often results in misdiagnosis. Extracardiac features should be considered for a timely AFD diagnosis [6,15][6][15] (Figure 1A). It is worth mentioning that disease expression varies across different ages (Figure 1B). The diagnostic workup of AFD should be based on a stepwise approach, including extracardiac and cardiac red flags, in order to recognize AFD as early as possible. (Figure 1C).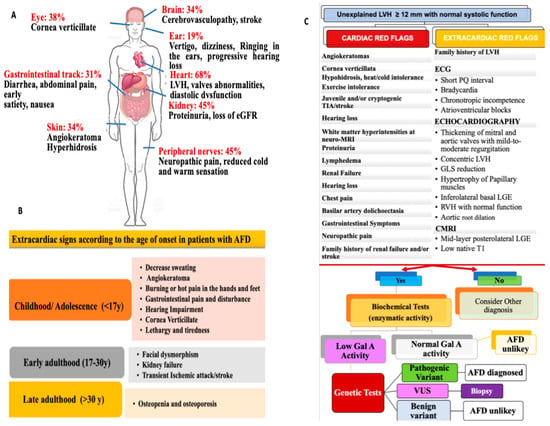
3. Cardiac Involvement
3.1. Pathophysiology
Myocardial accumulation of Gb3 is crucial in developing AFD cardiomyopathy, although the accumulation of GB3 alone does not explain all-spectrum cardiac manifestations. The initial deposition of Gb3 characterizes the early phase of the disease. This occurs especially in and around the atrioventricular (AV) node, leading to early conduction abnormalities in AFD. Similarly, the infiltrative process is considered the main underlying mechanism of abnormalities in conduction tissue, such as sinus node disease and AV block. Beyond Gb3 accumulation, other pathophysiological pathways of storage-triggered mechanisms might explain the whole spectrum of AFD cardiac disease and progression [2]. These mechanisms have been suggested to have a pivotal role in developing overt cardiac structural manifestations [18[18][28],28], including LVH and diastolic dysfunction [29,30][29][30]. Later, the progressive inflammation with the tumor-grown Factor-B-mediated extracellular matrix activation also correlates with myocardial fibrosis and remodeling in the advanced stage of cardiac involvement.3.2. Disease Manifestations: Patient Symptoms
Surveys and dedicated AFD registries lead to a unique opportunity to address cardiac clinical presentation [16,31,32,33][16][31][32][33]. Generally, cardiac symptoms have been reported with a higher prevalence in males than in females, increasing exponentially with age and disease progression for both genders [32]. Although cardiomyopathy is commonly asymptomatic during the early stage of AFD [31[31][32][34],32,34], index presentation as cardiac symptoms arises in almost 10% of patients. Otherwise, more than 60% experience HF, arrhythmias, angina, and syncope during the natural course of the disease [18,31,32][18][31][32]. The Fabry Outcome Survey (FOS) [35] and other selected AFD registries [5,16,31,32,36][5][16][31][32][36] have contributed to addressing cardiac manifestation in the affected population. Palpitations emerge as a clinical manifestation mostly related to supraventricular arrhythmias, with a higher prevalence in females than in males (21 vs. 15%) [32]. However, palpitations may also be a clinical manifestation of ventricular arrhythmias (VA), representing the most common life-threatening condition in AFD patients. As such, once AFD is suspected or diagnosed, the occurrence of palpitation should guide proper management.3.2.1. Electrophysiologic Abnormalities and Arrhythmias Burden
The electrocardiogram (ECG) remains an essential tool in the diagnostic assessment of AFD [22,42][22][37]. Various conduction abnormalities may be useful to detect cardiac involvement early. Therefore, the identification of ECG changes is crucial (Figure 2A). The early conduction manifestations are widely related to the accumulation of Gb3 affecting the conduction system [2]. Indeed, deposition of Gb3 around the AV node has been suggested as the earliest mechanism leading to an anomalous PR interval [43,44,45][38][39][40]. Notably, a short PR interval without a delta wave should increase the suspicion of AFD [44][39], often occurring before overt LVH development [14,45][14][40]. In this regard, interesting data has emerged from a study analysing conduction abnormalities in the ECGs of patients with newly diagnosed AFD without LVH. When these patients were compared with healthy controls, the PR interval was shorter in patients with early-stage FD (i.e., without LVH) than in the control group [45][40].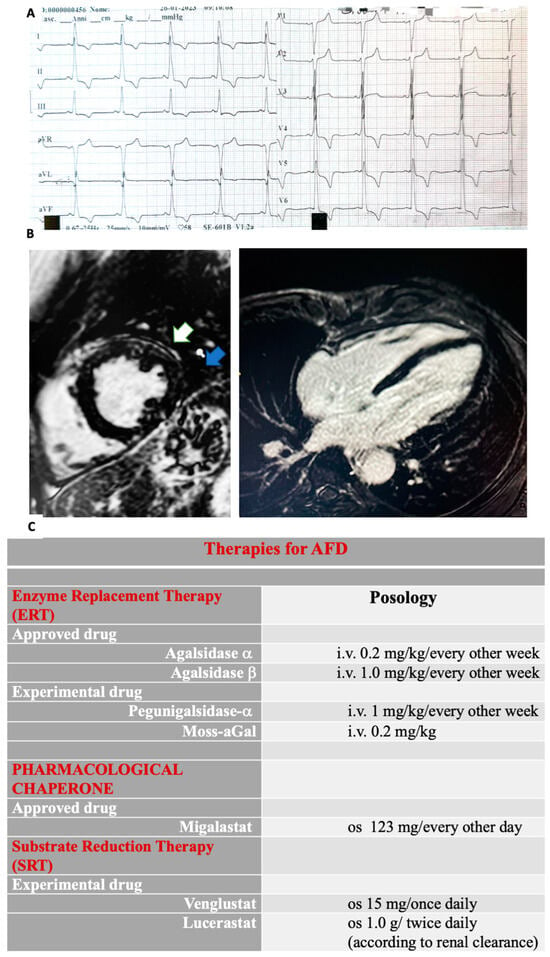
3.2.2. Echocardiographic Findings
3.2.3. Cardiac Magnetic Resonance Imaging Findings
CMRI is considered the gold standard for assessing LVH and myocardial fibrosis in AFD [61,62][49][50]. Remarkably, LGE in the subepicardial basal–mid-inferolateral wall is a hallmark of AFD cardiomyopathy. This is especially relevant in advanced-stage disease [62][50] (Figure 2B). T1 mapping is a well-established CMR technique used for assessing myocardial tissue characteristics and detecting myocardial edema, accumulation of intra-myocyte lipids, and expansion of extracellular volume, which may involve proteins or iron deposition [63,64][51][52]. It involves the measurement of the quantitative T1 signal originating from the myocardial tissue, which is then subjected to post-processing to generate a color-coded map representing the myocardium. Particularly, native T1 mapping evaluates the intrinsic myocardial longitudinal relaxation time without needing a contrast agent [63,64][51][52]. A low native T1 value is considered an indicator for identifying myocardial glycosphingolipid accumulation before the development of LVH, allowing a timely identification of cardiac impairment during a pre-hypertrophic phase [65][53]. Importantly, the reduction in T1 values during the pre-LVH stage has been correlated with a decrease in global longitudinal strain (GLS) [65][53]. Therefore, it has been demonstrated that a decreased T1 value within the context of LVH has a remarkable sensitivity and specificity for the recognition of AFD, enabling the differentiation of this condition from other hypertrophic forms where T1 values remain within the normal or elevated range, including hypertrophic cardiomyopathy (HCM), AL amyloidosis, hypertensive heart disease, severe aortic stenosis [66][54].4. Diagnostic Workup: The Roles of Genetic and Biochemical Testing, Biopsy, and Biomarkers in AFD
A definitive diagnosis should rely on genetic testing, enzyme activity, and tissue studies (whenever possible) showing Gb3 accumulation (Figure 1C). [1,14][1][14]. Specifically, biochemical measurements of α-Gal A activity in the blood and leukocytes occur through the detection of the plasma levels of the storage product GB3 and its degradation product (Lyso-GB3). For males with the classic form, in whom α-Gal A activity is severely reduced or absent, a biochemical test is often sufficient for diagnosis. In this clinical context, the assessment of α-Gal A enzyme activity should be performed as a first-line test. However, in rare cases, male patients might have residual α-Gal A activity, and this limits the diagnostic ability of the biochemical α-Gal A test. Genetic mutation research becomes necessary for these patients to diagnose [6]. Otherwise, for all AFD patients, genetic testing increases the diagnostic utility of the biochemical test. Indeed, different Gal A mutations are associated with a different spectrum of α-Gal A activity and disease manifestations. Particularly, non-sense, consensus splice site, and frameshift mutations are often related to lower or no α-Gal A enzyme activity. They are usually associated with the classic form of AFD. In contrast, missense mutations and rare cryptic splicing mutations can be associated with residual α-Gal A enzyme activity characterizing the late-onset phenotypes [6,15,74][6][15][55].5. Therapy
Disease-specific treatments for reducing CV events, such as enzyme replacement therapy (ERTs) [83,84][56][57] and the pharmacological chaperone migalastat [85,86][58][59] have been recently approved, while emerging molecules are developing. ERTs have dramatically improved the quality of life of AFD patients, reducing neuropathic pain, gastrointestinal symptoms, and CV events. ERTs prevent LVH development and favor LVH regression in the initial stage in patients with both classic and cardiac forms, exerting effects on cardiac structure, including a gradual reduction in the interventricular septum (IVS) thickening and a decrease in the left ventricle mass index (LVMi) [83,84,87,88][56][57][60][61]. Additionally, small-molecule chaperones’ use has been introduced for treating lysosomal storage disease [97][62]. Migalastat, an oral chaperone, favors enzymatic stabilization of the specific mutant α-Gal A variant (amenable forms) [85,86][58][59], seeming to reduce LVH, renal, and cardiovascular events [98][63] (Figure 2C). Particularly, an improvement in LVMi has been shown in the majority of patients treated with migalastat [98,99,100,101][63][64][65][66]. The randomized trials FAMOUS, FACETS, and ATTRACT demonstrated a significant reduction in LVMi after 24 and 30 months of therapy [98,99,102,103][63][64][67][68].References
- Marian, A.J. Challenges in the diagnosis of anderson-fabry disease. J. Am. Coll. Cardiol. 2016, 68, 1051–1053.
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac involvement in fabry disease: Jacc review topic of the week. J. Am. Coll. Cardiol. 2021, 77, 922–936.
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with fabry disease: Findings from the fabry registry. Genet. Med. 2009, 11, 790–796.
- Vardarli, I.; Weber, M.; Rischpler, C.; Führer, D.; Herrmann, K.; Weidemann, F. Fabry cardiomyopathy: Current treatment and future options. J. Clin. Med. 2021, 10, 2750.
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with fabry disease receiving agalsidase β: Data from the fabry registry. J. Med. Genet. 2016, 53, 495–502.
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427.
- Citro, R.; Prota, C.; Ferraioli, D.; Iuliano, G.; Bellino, M.; Radano, I.; Silverio, A.; Migliarino, S.; Polito, M.V.; Ruggiero, A.; et al. Importance of echocardiography and clinical “red flags” in guiding genetic screening for fabry disease. Front. Cardiovasc. Med. 2022, 9, 838200.
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in fabry disease: A systematic review of risk factors in clinical practice. Europace 2018, 20, f153–f161.
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. Jama 1999, 281, 249–254.
- Inoue, T.; Hattori, K.; Ihara, K.; Ishii, A.; Nakamura, K.; Hirose, S. Newborn screening for fabry disease in japan: Prevalence and genotypes of fabry disease in a pilot study. J. Hum. Genet. 2013, 58, 548–552.
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40.
- Wozniak, M.A.; Kittner, S.J.; Tuhrim, S.; Cole, J.W.; Stern, B.; Dobbins, M.; Grace, M.E.; Nazarenko, I.; Dobrovolny, R.; McDade, E.; et al. Frequency of unrecognized fabry disease among young european-american and african-american men with first ischemic stroke. Stroke 2010, 41, 78–81.
- Linhart, A.; Elliott, P.M. The heart in anderson-fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535.
- Favalli, V.; Disabella, E.; Molinaro, M.; Tagliani, M.; Scarabotto, A.; Serio, A.; Grasso, M.; Narula, N.; Giorgianni, C.; Caspani, C.; et al. Genetic screening of anderson-fabry disease in probands referred from multispecialty clinics. J. Am. Coll. Cardiol. 2016, 68, 1037–1050.
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435.
- Mehta, A.; Clarke, J.T.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of fabry disease: Changing pattern of causes of death in fos—fabry outcome survey. J. Med. Genet. 2009, 46, 548–552.
- Patel, V.; O’Mahony, C.; Hughes, D.; Rahman, M.S.; Coats, C.; Murphy, E.; Lachmann, R.; Mehta, A.; Elliott, P.M. Clinical and genetic predictors of major cardiac events in patients with anderson-fabry disease. Heart 2015, 101, 961–966.
- Akhtar, M.M.; Elliott, P.M. Anderson-fabry disease in heart failure. Biophys. Rev. 2018, 10, 1107–1119.
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An atypical variant of fabry’s disease in men with left ventricular hypertrophy. N. Engl. J. Med. 1995, 333, 288–293.
- von Scheidt, W.; Eng, C.M.; Fitzmaurice, T.F.; Erdmann, E.; Hübner, G.; Olsen, E.G.; Christomanou, H.; Kandolf, R.; Bishop, D.F.; Desnick, R.J. An atypical variant of fabry’s disease with manifestations confined to the myocardium. N. Engl. J. Med. 1991, 324, 395–399.
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with fabry disease. Clin. Genet. 2016, 89, 44–54.
- Vitale, G.; Ditaranto, R.; Graziani, F.; Tanini, I.; Camporeale, A.; Lillo, R.; Rubino, M.; Panaioli, E.; Di Nicola, F.; Ferrara, V.; et al. Standard ecg for differential diagnosis between anderson-fabry disease and hypertrophic cardiomyopathy. Heart 2022, 108, 54–60.
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry disease: Prevalence of affected males and heterozygotes with pathogenic gla mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J Med Genet 2018, 55, 261–268.
- Stankowski, K.; Figliozzi, S.; Battaglia, V.; Catapano, F.; Francone, M.; Monti, L. Fabry disease: More than a phenocopy of hypertrophic cardiomyopathy. J. Clin. Med. 2023, 12, 7061.
- Militaru, S.; Jurcuț, R.; Adam, R.; Roşca, M.; Ginghina, C.; Popescu, B.A. Echocardiographic features of fabry cardiomyopathy-comparison with hypertrophy-matched sarcomeric hypertrophic cardiomyopathy. Echocardiography 2019, 36, 2041–2049.
- Karur, G.R.; Robison, S.; Iwanochko, R.M.; Morel, C.F.; Crean, A.M.; Thavendiranathan, P.; Nguyen, E.T.; Mathur, S.; Wasim, S.; Hanneman, K. Use of myocardial t1 mapping at 3.0 t to differentiate anderson-fabry disease from hypertrophic cardiomyopathy. Radiology 2018, 288, 398–406.
- Wechalekar, A.D.; Fontana, M.; Quarta, C.C.; Liedtke, M. Al amyloidosis for cardiologists: Awareness, diagnosis, and future prospects: Jacc: Cardiooncology state-of-the-art review. JACC CardioOncol 2022, 4, 427–441.
- Azevedo, O.; Cordeiro, F.; Gago, M.F.; Miltenberger-Miltenyi, G.; Ferreira, C.; Sousa, N.; Cunha, D. Fabry disease and the heart: A comprehensive review. Int. J. Mol. Sci. 2021, 22, 4434.
- Frustaci, A.; Morgante, E.; Russo, M.A.; Scopelliti, F.; Grande, C.; Verardo, R.; Franciosa, P.; Chimenti, C. Pathology and function of conduction tissue in fabry disease cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2015, 8, 799–805.
- Ikari, Y.; Kuwako, K.; Yamaguchi, T. Fabry’s disease with complete atrioventricular block: Histological evidence of involvement of the conduction system. Br. Heart J. 1992, 68, 323–325.
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia de Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the fabry outcome survey. Eur. J. Clin. Invest. 2004, 34, 236–242.
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M. Cardiac manifestations of anderson-fabry disease: Results from the international fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235.
- Wu, J.C.; Ho, C.Y.; Skali, H.; Abichandani, R.; Wilcox, W.R.; Banikazemi, M.; Packman, S.; Sims, K.; Solomon, S.D. Cardiovascular manifestations of fabry disease: Relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase a activity. Eur. Heart J. 2010, 31, 1088–1097.
- Selthofer-Relatic, K. Time of anderson-fabry disease detection and cardiovascular presentation. Case Rep. Cardiol. 2018, 2018, 6131083.
- Clarke, J.T.; Giugliani, R.; Sunder-Plassmann, G.; Elliott, P.M.; Pintos-Morell, G.; Hernberg-Ståhl, E.; Malmenäs, M.; Beck, M. Impact of measures to enhance the value of observational surveys in rare diseases: The fabry outcome survey (fos). Value Health 2011, 14, 862–866.
- Eng, C.M.; Fletcher, J.; Wilcox, W.R.; Waldek, S.; Scott, C.R.; Sillence, D.O.; Breunig, F.; Charrow, J.; Germain, D.P.; Nicholls, K.; et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the fabry registry. J. Inherit. Metab. Dis. 2007, 30, 184–192.
- Namdar, M. Electrocardiographic changes and arrhythmia in fabry disease. Front. Cardiovasc. Med. 2016, 3, 7.
- Kampmann, C.; Wiethoff, C.M.; Whybra, C.; Baehner, F.A.; Mengel, E.; Beck, M. Cardiac manifestations of anderson-fabry disease in children and adolescents. Acta Paediatr. 2008, 97, 463–469.
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Heliö, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the esc working group on myocardial and pericardial diseases. Eur. Heart J. 2013, 34, 1448–1458.
- Namdar, M.; Steffel, J.; Vidovic, M.; Brunckhorst, C.B.; Holzmeister, J.; Lüscher, T.F.; Jenni, R.; Duru, F. Electrocardiographic changes in early recognition of fabry disease. Heart 2011, 97, 485–490.
- Yeung, D.F.; Sirrs, S.; Tsang, M.Y.C.; Gin, K.; Luong, C.; Jue, J.; Nair, P.; Lee, P.K.; Tsang, T.S.M. Echocardiographic assessment of patients with fabry disease. J. Am. Soc. Echocardiogr. 2018, 31, 639–649.e632.
- Tower-Rader, A.; Jaber, W.A. Multimodality imaging assessment of fabry disease. Circ. Cardiovasc. Imaging 2019, 12, e009013.
- Chimenti, C.; Russo, M.A.; Frustaci, A. Atrial biopsy evidence of fabry disease causing lone atrial fibrillation. Heart 2010, 96, 1782–1783.
- Linhart, A.; Palecek, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupetová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with fabry’s disease. Am. Heart J. 2000, 139, 1101–1108.
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of anderson-fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002, 105, 1407–1411.
- Calcagnino, M.; O’Mahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Janagarajan, K.; Mehta, A.; Hughes, D.; Murphy, E.; Lachmann, R.; et al. Exercise-induced left ventricular outflow tract obstruction in symptomatic patients with anderson-fabry disease. J. Am. Coll. Cardiol. 2011, 58, 88–89.
- Mehta, A.; Beck, M.; Sunder-Plassmann, G. Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006.
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Störk, S.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. The variation of morphological and functional cardiac manifestation in fabry disease: Potential implications for the time course of the disease. Eur. Heart J. 2005, 26, 1221–1227.
- Deva, D.P.; Hanneman, K.; Li, Q.; Ng, M.Y.; Wasim, S.; Morel, C.; Iwanochko, R.M.; Thavendiranathan, P.; Crean, A.M. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in anderson-fabry disease. J. Cardiovasc. Magn. Reson. 2016, 18, 14.
- Weidemann, F.; Sommer, C.; Duning, T.; Lanzl, I.; Möhrenschlager, M.; Naleschinski, D.; Arning, K.; Baron, R.; Niemann, M.; Breunig, F.; et al. Department-related tasks and organ-targeted therapy in fabry disease: An interdisciplinary challenge. Am. J. Med. 2010, 123, e651–e658.
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of t1, t2, t2* and extracellular volume: A consensus statement by the society for cardiovascular magnetic resonance (SCMR) endorsed by the european association for cardiovascular imaging (EACVI). J. Cardiovasc. Magn. Reson. 2017, 19, 75.
- Schelbert, E.B.; Messroghli, D.R. State of the art: Clinical applications of cardiac t1 mapping. Radiology 2016, 278, 658–676.
- Augusto, J.B.; Johner, N.; Shah, D.; Nordin, S.; Knott, K.D.; Rosmini, S.; Lau, C.; Alfarih, M.; Hughes, R.; Seraphim, A.; et al. The myocardial phenotype of fabry disease pre-hypertrophy and pre-detectable storage. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 790–799.
- Ho, C.Y.; Abbasi, S.A.; Neilan, T.G.; Shah, R.V.; Chen, Y.; Heydari, B.; Cirino, A.L.; Lakdawala, N.K.; Orav, E.J.; González, A.; et al. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ. Cardiovasc. Imaging 2013, 6, 415–422.
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30.
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and efficacy of recombinant human alpha-galactosidase a replacement therapy in fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16.
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in fabry disease: A randomized controlled trial. Jama 2001, 285, 2743–2749.
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 2016, 375, 545–555.
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438.
- Schiffmann, R.; Murray, G.J.; Treco, D.; Daniel, P.; Sellos-Moura, M.; Myers, M.; Quirk, J.M.; Zirzow, G.C.; Borowski, M.; Loveday, K.; et al. Infusion of alpha-galactosidase a reduces tissue globotriaosylceramide storage in patients with fabry disease. Proc. Natl. Acad. Sci. USA 2000, 97, 365–370.
- Spinelli, L.; Pisani, A.; Sabbatini, M.; Petretta, M.; Andreucci, M.V.; Procaccini, D.; Lo Surdo, N.; Federico, S.; Cianciaruso, B. Enzyme replacement therapy with agalsidase beta improves cardiac involvement in fabry’s disease. Clin. Genet. 2004, 66, 158–165.
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031–1045.
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in fabry disease: 18-month results from the randomised phase iii attract study. J. Med. Genet. 2017, 54, 288–296.
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Eveslage, M.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; et al. Treatment of fabry disease management with migalastat-outcome from a prospective 24 months observational multicenter study (famous). Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 272–281.
- Narita, I.; Ohashi, T.; Sakai, N.; Hamazaki, T.; Skuban, N.; Castelli, J.P.; Lagast, H.; Barth, J.A. Efficacy and safety of migalastat in a japanese population: A subgroup analysis of the attract study. Clin. Exp. Nephrol. 2020, 24, 157–166.
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; Üçeyler, N.; et al. Treatment of fabry’s disease with migalastat: Outcome from a prospective observational multicenter study (famous). Clin. Pharmacol. Ther. 2020, 108, 326–337.
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in fabry disease: 30-month results from the open-label extension of the randomized, phase 3 attract study. Mol. Genet. Metab. 2020, 131, 219–228.
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997.