Importantly, there is a certain degree of overlap between HCM and AFD, which may be considered a phenocopy of HCM. Therefore, some individuals with AFD are often misdiagnosed with HCM
[22].
A prevalence of GLA gene mutations has been reported in approximately 1% of HCM patients, particularly in those with late-onset cardiac variants
[23]. Similarly, among individuals undergoing surgical myectomy, genetic analysis revealed a GLA mutation in 1.3% of the patients. In light of these findings, a systematic screening for AFD in patients exhibiting the HCM phenotype should be performed. In cases of non-obstructive HCM, the LVH distribution can vary
[24].
In HCM, late gadolinium enhancement (LGE) and impaired regional strain are typically observed in the most hypertrophic segments. In a study on 40 patients with LVH, including both AFD and HCM patients and matched for the degree of LVH and age, the FD group exhibited a lower left ventricular ejection fraction (LVEF), more reduced regional longitudinal strain (LS) in the inferolateral LV wall, and a more impaired right ventricular (RV) free wall LS
[25].
Moreover, a higher LA enlargement degree and worse left atrial (LA) strain have been reported in HCM compared to AFD. Furthermore, a septal native T1 value < 1220 ms has been considered a helpful finding in differentiating FD from HCM with an accuracy of 95%
[26].
Amyloidosis is a systemic disorder involving more than one organ, including the heart, kidneys, liver, and autonomic nervous system.
Immunoglobulin light chain (AL) amyloidosis and transthyretin (ATTR) amyloidosis have been recognized as the two predominant types of infiltrating amyloid. Hereditary transthyretin (TTR) amyloidosis is a result of a genetic mutation that predisposes individuals to the instability of the tetrameric structure of transthyretin
[27].
Specific diagnostic tools, including bone scintigraphy, light chain assays, and tissue biopsies, are essential in confirming CA diagnosis. In native CA, T1 values and ECV increases have been described, with a global subendocardial or transmural LGE pattern
[27].
In the context of echocardiographic assessment of CA, concentric LVH parameters such as posterior wall thickness (PWTd), interventricular septum thickness (IVSd), and relative wall thickness (RWT), along with a decrease in LS are considered characteristics
[27].
3. Cardiac Involvement
3.1. Pathophysiology
Myocardial accumulation of Gb3 is crucial in developing AFD cardiomyopathy, although the accumulation of GB3 alone does not explain all-spectrum cardiac manifestations. The initial deposition of Gb3 characterizes the early phase of the disease. This occurs especially in and around the atrioventricular (AV) node, leading to early conduction abnormalities in AFD. Similarly, the infiltrative process is considered the main underlying mechanism of abnormalities in conduction tissue, such as sinus node disease and AV block. Beyond Gb3 accumulation, other pathophysiological pathways of storage-triggered mechanisms might explain the whole spectrum of AFD cardiac disease and progression
[2].
These mechanisms have been suggested to have a pivotal role in developing overt cardiac structural manifestations
[18][28][18,28], including LVH and diastolic dysfunction
[29][30][29,30]. Later, the progressive inflammation with the tumor-grown Factor-B-mediated extracellular matrix activation also correlates with myocardial fibrosis and remodeling in the advanced stage of cardiac involvement.
3.2. Disease Manifestations: Patient Symptoms
Surveys and dedicated AFD registries lead to a unique opportunity to address cardiac clinical presentation
[16][31][32][33][16,31,32,33]. Generally, cardiac symptoms have been reported with a higher prevalence in males than in females, increasing exponentially with age and disease progression for both genders
[32]. Although cardiomyopathy is commonly asymptomatic during the early stage of AFD
[31][32][34][31,32,34], index presentation as cardiac symptoms arises in almost 10% of patients. Otherwise, more than 60% experience HF, arrhythmias, angina, and syncope during the natural course of the disease
[18][31][32][18,31,32]. The Fabry Outcome Survey (FOS)
[35] and other selected AFD registries
[5][16][31][32][36][5,16,31,32,36] have contributed to addressing cardiac manifestation in the affected population.
Palpitations emerge as a clinical manifestation mostly related to supraventricular arrhythmias, with a higher prevalence in females than in males (21 vs. 15%)
[32]. However, palpitations may also be a clinical manifestation of ventricular arrhythmias (VA), representing the most common life-threatening condition in AFD patients. As such, once AFD is suspected or diagnosed, the occurrence of palpitation should guide proper management.
3.2.1. Electrophysiologic Abnormalities and Arrhythmias Burden
The electrocardiogram (ECG) remains an essential tool in the diagnostic assessment of AFD
[22][37][22,42]. Various conduction abnormalities may be useful to detect cardiac involvement early. Therefore, the identification of ECG changes is crucial (
Figure 2A). The early conduction manifestations are widely related to the accumulation of Gb3 affecting the conduction system
[2]. Indeed, deposition of Gb3 around the AV node has been suggested as the earliest mechanism leading to an anomalous PR interval
[38][39][40][43,44,45]. Notably, a short PR interval without a delta wave should increase the suspicion of AFD
[39][44], often occurring before overt LVH development
[14][40][14,45]. In this regard, interesting data has emerged from a study analysing conduction abnormalities in the ECGs of patients with newly diagnosed AFD without LVH. When these patients were compared with healthy controls, the PR interval was shorter in patients with early-stage FD (i.e., without LVH) than in the control group
[40][45].
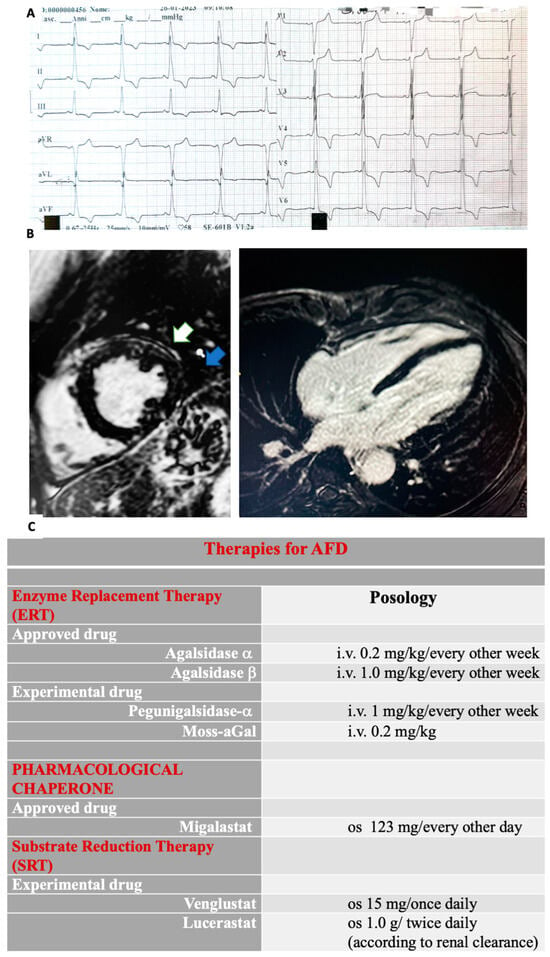
Figure 2. (
A) Giant negative T-waves are shown in the ECG. (
B) Cardiac magnetic resonance imaging (CMRI) short axis (
A) and four-chamber (
B) mid-basal inferolateral late gadolinium enhancement (LGE) in a patient with Anderson–Fabry disease. In the mid-antero-lateral wall, LGE is shown (white arrow). Intramyocardial LGE is shown in the mid-inferior-lateral wall (blue arrow). Recently, T1 mapping analysis has been proposed as a reliable tool for diagnosing AFD, demonstrating high sensitivity and specificity to discriminate this cardiomyopathy within a wide spectrum of conditions involving myocardial hypertrophy. (
C) A timely multidisciplinary treatment consisting of both an FD-specific and a cardiovascular approach is crucial in order to avoid progression and irreversible systemic failure. ERTs and chaperone migalastat represent the approved disease-specific pharmacological strategies. Two different ERTs are used to treat AFD: algasidase alpha and algasidase beta. They both contain recombinant human a-Gal A and exhibit identical biochemical profiles. Nevertheless, different dosage regimens are used. ERTs have been shown to improve symptoms and reduce CV events and disease progression, particularly in both classic and cardiac forms, while in late-onset and advanced cardiac AFD cases, their effectiveness is poor. Since ERTs consist of recombinant human protein (a-Gal A), the development of neutralizing antibodies directed against the enzyme has been reported, particularly in males with classic AFD. Remarkably, the presence of antibodies against the enzyme decreases therapy efficacy. Algasidase alpha and algasidase beta have been approved in Europe and Canada, while in the USA, only algasidase beta use is allowed. Different studies have shown similar efficacy with no differences in the clinical event rates, although patients treated with algasidase beta were more likely to have a higher reduction in left ventricular mass (LVH). Moreover, less development of antibodies has been associated with algasidase beta than with algasidase alpha. In addition, chaperone therapy has been introduced to treat lysosomal storage disease. A small-molecule chaperone interacts with a mutant enzyme favoring its correct conformation, stability, and functioning. Migalastat, an oral pharmacological chaperone, has been recently proposed as an alternative to intravenous ERT in AFD. Its pharmacological action consists of stabilizing specific mutant (amenable) forms of α-Gal A in order to facilitate normal lysosomal trafficking. Moreover, other new therapies include second-generation ERTs and substrate reduction therapies (SRT). Gene and mRNA therapies are currently developing. Pegunigalsidase-a is a novel pegylated form of a-Gal A. Characteristically, its circulatory half-life is long-lasting. Moreover, heart and kidney uptakes are higher compared to current ERTs. Moss-aGalactosidase A (moss-aGal) is a moss-derived variant of human α-galactosidase. SRTs are another object of studies (venglustat is currently in phase II, whereas lucerastat is in phase III of clinical trials).
3.2.2. Echocardiographic Findings
Echocardiography is an effective noninvasive tool for assessing structural and functional cardiac involvement in AFD
[15][41][42][15,49,50]. LVH occurs in more than 50% and 20% of males and females, representing a key feature in AFD. The concentric pattern is the most common structural abnormality, although eccentric, asymmetric, and distal distributions have also been described
[33][43][33,47]. Although LVH manifestation is usually delayed in females, a similar incidence has been reported in both genders
[44][51].
AFD often mimics hypertrophic cardiomyopathy (HCM) without left ventricular outflow tract obstruction (LVOTO). However, LVOTO, along with papillary muscle hypertrophy, may occur
[44][45][51,52]. Indeed, resting LVOTO is rare, but it may be provokable during exercise in about 50% of patients with LVH related to AFD
[46][53].
Right ventricular (RV) hypertrophy may also develop in nearly 25% of patients, with similar prevalence for both genders
[33]. Ongoing valvular abnormalities may occur in both right- and left-sided valves, although mitral or aortic involvement appears to be more relevant
[47][54].
Pulsed wave Doppler and tissue Doppler imaging (TDI) are useful to assess subclinical diastolic and systolic dysfunction, which may occur before the development of overt LVH
[48][56]. Diastolic dysfunction is related to increased ventricular stiffness and impaired relaxation due to intracellular Gb3 deposition and myocardial fibrosis TDI. The longitudinal or circumferential strain rate may reveal subclinical cardiac involvement before the onset of LVH or systolic/diastolic impairment. However, TDI remains unspecific and poor in discriminating AFD from other cardiomyopathies.
3.2.3. Cardiac Magnetic Resonance Imaging Findings
CMRI is considered the gold standard for assessing LVH and myocardial fibrosis in AFD
[49][50][61,62]. Remarkably, LGE in the subepicardial basal–mid-inferolateral wall is a hallmark of AFD cardiomyopathy. This is especially relevant in advanced-stage disease
[50][62] (
Figure 2B). T1 mapping is a well-established CMR technique used for assessing myocardial tissue characteristics and detecting myocardial edema, accumulation of intra-myocyte lipids, and expansion of extracellular volume, which may involve proteins or iron deposition
[51][52][63,64].
It involves the measurement of the quantitative T1 signal originating from the myocardial tissue, which is then subjected to post-processing to generate a color-coded map representing the myocardium. Particularly, native T1 mapping evaluates the intrinsic myocardial longitudinal relaxation time without needing a contrast agent
[51][52][63,64].
A low native T1 value is considered an indicator for identifying myocardial glycosphingolipid accumulation before the development of LVH, allowing a timely identification of cardiac impairment during a pre-hypertrophic phase
[53][65]. Importantly, the reduction in T1 values during the pre-LVH stage has been correlated with a decrease in global longitudinal strain (GLS)
[53][65].
Therefore, it has been demonstrated that a decreased T1 value within the context of LVH has a remarkable sensitivity and specificity for the recognition of AFD, enabling the differentiation of this condition from other hypertrophic forms where T1 values remain within the normal or elevated range, including hypertrophic cardiomyopathy (HCM), AL amyloidosis, hypertensive heart disease, severe aortic stenosis
[54][66].
4. Diagnostic Workup: The Roles of Genetic and Biochemical Testing, Biopsy, and Biomarkers in AFD
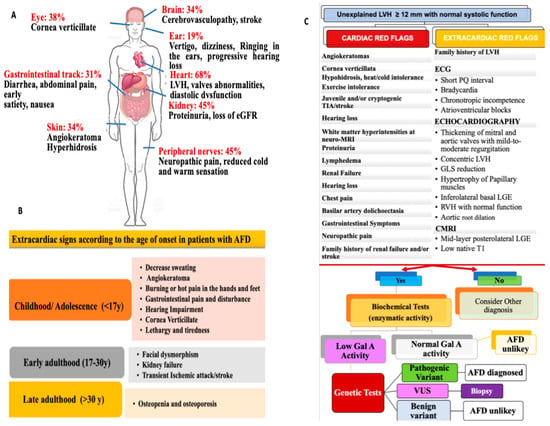
A definitive diagnosis should rely on genetic testing, enzyme activity, and tissue studies (whenever possible) showing Gb3 accumulation (
Figure 1C).
[1][14][1,14].
Specifically, biochemical measurements of α-Gal A activity in the blood and leukocytes occur through the detection of the plasma levels of the storage product GB3 and its degradation product (Lyso-GB3). For males with the classic form, in whom α-Gal A activity is severely reduced or absent, a biochemical test is often sufficient for diagnosis. In this clinical context, the assessment of α-Gal A enzyme activity should be performed as a first-line test. However, in rare cases, male patients might have residual α-Gal A activity, and this limits the diagnostic ability of the biochemical α-Gal A test. Genetic mutation research becomes necessary for these patients to diagnose
[6].
Otherwise, for all AFD patients, genetic testing increases the diagnostic utility of the biochemical test. Indeed, different Gal A mutations are associated with a different spectrum of α-Gal A activity and disease manifestations. Particularly, non-sense, consensus splice site, and frameshift mutations are often related to lower or no α-Gal A enzyme activity. They are usually associated with the classic form of AFD. In contrast, missense mutations and rare cryptic splicing mutations can be associated with residual α-Gal A enzyme activity characterizing the late-onset phenotypes
[6][15][55][6,15,74].
5. Therapy
Disease-specific treatments for reducing CV events, such as enzyme replacement therapy (ERTs)
[56][57][83,84] and the pharmacological chaperone migalastat
[58][59][85,86] have been recently approved, while emerging molecules are developing. ERTs have dramatically improved the quality of life of AFD patients, reducing neuropathic pain, gastrointestinal symptoms, and CV events. ERTs prevent LVH development and favor LVH regression in the initial stage in patients with both classic and cardiac forms, exerting effects on cardiac structure, including a gradual reduction in the interventricular septum (IVS) thickening and a decrease in the left ventricle mass index (LVMi)
[56][57][60][61][83,84,87,88].
Additionally, small-molecule chaperones’ use has been introduced for treating lysosomal storage disease
[62][97]. Migalastat, an oral chaperone, favors enzymatic stabilization of the specific mutant α-Gal A variant (amenable forms)
[58][59][85,86], seeming to reduce LVH, renal, and cardiovascular events
[63][98] (
Figure 2C). Particularly, an improvement in LVMi has been shown in the majority of patients treated with migalastat
[63][64][65][66][98,99,100,101]. The randomized trials FAMOUS, FACETS, and ATTRACT demonstrated a significant reduction in LVMi after 24 and 30 months of therapy
[63][64][67][68][98,99,102,103].